


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




景小蔓 张洋洋 金 英 俞菁娴 高行健 张嘉会 钟 清 刘志红 张昌明
DOI:10.3969/j.issn.1006-298X.2024.05.006
[基金项目] 国家重点研发计划(2021YFC2501302);国家自然科学基金(82170739);东部战区总医院院管课题(2023LCYYQH028);江苏省科技资源统筹服务平台肾脏疾病样本库运行经费(BM2015004-1)
[作者单位] 东南大学医学院(东部战区总医院)硕士研究生(景小蔓) 国家肾脏疾病临床医学研究中心(南京,210016)
[通信作者] 张昌明(E-mail:zhangchmingg@hotmail.com)
摘 要 目的:分析肿瘤坏死因子α诱导蛋白3(TNFAIP3)基因突变导致的狼疮性肾炎(LN)患者的表型特征。方法:收集2例经全外显子测序检测出携带TNFAIP3基因突变的LN患者的临床和病理资料,通过Sanger测序验证突变,并结合文献检索总结此类患者的表型和致病变异谱特征。结果:病例1,女性,19岁起病,肾脏受累表现为中等量蛋白尿及镜下血尿,肾活检提示LN-Ⅳ+V型,有反复发热、面部红斑、关节疼痛、溶血性贫血和癫痫发作等肾外表现。全外显子测序发现TNFAIP3基因c.C811T(p.R271*)变异,为新发变异。病例2,男性,29岁起病,肾脏表现为大量蛋白尿及镜下血尿,肾活检提示LN-Ⅳ型。有皮疹、脱发、关节疼痛、口干、眼干、过敏等肾外表现。患者母亲有复发性无痛性口腔溃疡。全外显子测序发现,该患者携带TNFAIP3基因c.634+2T>C(p.D212Gfs37*)变异,其母亲也携带该突变。文献检索到TNFAIP3基因突变导致的LN共14例(包括本研究2例),检出13个TNFAIP3基因杂合突变。14例患者中女性(12例,85.71%)为主,多儿童期起病,仅2例成人期起病。皮疹,口腔和(或)生殖器溃疡,反复发热,和关节炎是最常见的肾外表现,10例(71.43%)患者合并多种自身免疫性疾病。多数患者在免疫抑制剂或生物制剂治疗后症状得到有效控制。结论:TNFAIP3基因突变导致的LN表现复杂。对合并多种自身免疫表现的LN患者,应详细询问家族史,及时完善基因测序,以帮助早期诊断和指导个体化治疗。
关键词 狼疮性肾炎 肿瘤坏死因子α诱导蛋白3 A20单倍剂量不足 自身免疫
JING Xiaoman, ZHANG Yangyang, JIN Ying, YU Jingxian, GAO Xingjian, ZHANG Jiahui, ZHONG Qing, LIU Zhihong, ZHANG Changming
National Clinical Research Center for Kidney Diseases, Jinling Hospital, Southeast University School of Medicine, Nanjing 210016, China
Corresponding author:ZHANG Changming(E-mail:zhangchmingg@hotmail.com)
ABSTRACT Objective:To analyze the phenotypic characteristics of patients with lupus nephritis (LN) caused by mutation of TNF-alpha-induced protein 3(TNFAIP3) gene.
Methodology:Clinical and pathological data were collected from 2 LN patients with TNFAIP3 mutations identified through whole-exome sequencing. Validation of the mutations was performed using Sanger sequencing. A comprehensive review of the literature was conducted to summarize the clinical characteristics and pathogenic mutation spectrum.
Results:Patient 1, a female who presented with symptoms at the age of 19, showed kidney involvement characterized by moderate proteinuria and microscopic hematuria. Kidney biopsy indicated LN-Ⅳ+V. Extra-kidney manifestations included recurrent fever, facial erythema, joint pain, hemolytic anemia, and seizures. Whole-exome sequencing revealed a de novo TNFAIP3 gene mutation c.C811T (p.R271*). Patient 2, a male, onset at 29 years, displayed significant proteinuria and microscopic hematuria, with kidney biopsy indicating LN-Ⅳ. Extra-kidney symptoms included rash, hair loss, joint pain, dry mouth, dry eyes, and allergies. His mother had recurrent painless oral ulcers. Whole-exome sequencing identified a TNFAIP3 gene mutation c.634+2T>C (p.D212Gfs*37) inherited from his mother. Literature search found that 14 cases of TNFAIP3 mutation combined with LN were reported, including this study. A total of 13 TNFAIP3 mutations were detected, all of which were heterozygous. Among these patients, females were predominant (12 cases, 85.71%), with onset mostly in childhood; only 2 cases had adult onset. Rash, oral and/or genital ulcers, recurrent fever, and arthritis were the most common extra-kidney manifestations. Ten (71.43%) had multiple autoimmune diseases. The symptoms are effectively controlled after immunosuppressive or biologic treatment in most patients.
Conclusion:LN expression caused by TNFAIP3 gene mutations is complex. For LN patients with multiple autoimmune manifestations, a detailed family history and genetic sequencing should be performed in time to help early diagnosis and personalized treatment.
Key words lupus nephritis TNFAIP3 haploinsufficiency of A20 autoimmunity
肿瘤坏死因子α诱导蛋白3(TNFAIP3)基因编码A20蛋白,在调控免疫反应、维持免疫系统的平衡以及保护机体免受炎症伤害方面扮演着重要角色。TNFAIP3基因功能缺失性突变引发一种罕见的常染色体显性遗传的自身炎症性疾病,称为A20单倍剂量不足(HA20, OMIM 616744)[1]。此类患者常在幼年时期就出现症状,包括发热、黏膜溃疡、葡萄膜炎、关节炎和皮肤损伤等。HA20患者症状与白塞病、类风湿性关节炎、炎症性肠病等广泛重叠,易被误诊。TNFAIP3基因突变时,核因子κB(NF-κB)通路持续活化会导致炎症细胞因子过量产生,促进炎症和自身免疫反应的风险,从而影响肾脏等重要器官[2]。部分HA20患者出现狼疮样表现和免疫学异常[3-4]。本文报道2例成人起病以系统性红斑狼疮(SLE)及狼疮性肾炎(LN)为主要表现的TNFAIP3基因突变的患者,并通过文献复习总结TNFAIP3基因突变导致的LN患者的表型及致病变异谱特征。
研究对象 2003年及2015年于国家肾脏疾病临床医学研究中心就诊的2例LN患者,符合1997年《美国风湿病学会修订的SLE分类标准》,肾活检病理符合LN,行全外显子测序发现,携带TNFAIP3基因变异,收集其临床及病理资料,并对直系亲属进行家系验证。全外显子组测序及变异的致病性分析参照本中心前期建立的方法进行[5]。
Sanger测序 分别对2个家系基因组DNA进行聚合酶链式反应(PCR)扩增。针对候选变异位点,采用《Snapgene》软件设计PCR引物,病例1:F:AGCCAGGATTCAAACTCAGG,R:GTTTCCCATTGG CAGAAACT。病例2:F:TCCCCAACTTTTGAGTTTGC, R:GGCAAGTAAATTCCACCCACT。
剪切位点变异对可变剪切的影响 采用PAXgene血液RNA管采集病例2的外周血,PAXgene血液RNA提取试剂盒(货号#762174)提取总RNA后,进行逆转录及PCR,Sanger测序检测基因序列变化。
文献检索 以"TNFAIP3" "HA20" "A20 haploinsufficiency" "A20单倍剂量不足" 等为关键词,检索截至2024年1月的PubMed、Web of Science、CNKI、万方等数据库,共检索到45篇文献。
病例介绍 病例1,女性,1996年(19岁)起反复出现多关节肿痛和运动障碍,累及手指、足趾、踝、肘和膝关节,伴雷诺现象,2000年起间断发热,对雷公藤多苷和非甾体抗炎药反应良好。2001年因“产后宫内感染、感染性休克、高热”住院,发现重度溶血性贫血。予抗生素、大量输血后体温降至37.3℃,血红蛋白回升。2003年出现眼睑水肿,伴高热,蛋白尿,低蛋白血症,血培养示表皮葡萄球菌生长,检测到狼疮细胞。经万古霉素、罗氏芬及甲泼尼龙治疗后仍高热。后至我院检查示尿蛋白定量3.34 g/d,溶血性贫血(血红蛋白45g/L),白细胞2.6×109/L,血清肌酐(SCr)正常,抗核抗体(ANA)、抗双链DNA(dsDNA)、抗心磷脂抗体、类风湿因子阳性,补体C3 0.389 g/L、C4 0.022 g/L。胸部CT提示双侧胸腔积液、双侧腋窝多发淋巴结,考虑狼疮肺炎。肾活检符合LN-V+Ⅳ型[活动性指数(AI)14分,慢性化指数(CI)1分]。住院期间出现两次癫痫发作,MRI检查提示急性脑梗,抗凝及丙戊酸钠治疗后癫痫未再发作。
经甲泼尼龙1.5 g冲击,泼尼松联合吗替麦考酚酯治疗,尿检完全缓解,补体恢复正常,自身抗体滴度下降,后泼尼松联合雷公藤多苷或硫唑嘌呤维持。随访期间病情相对稳定,尿检基本阴性,但劳累及感染后曾复发,表现为尿蛋白转阳,血小板及补体降低,伴低热和皮肤瘀斑。2011年重复肾活检示,LN-Ⅳ+V型(AI 12分,CI 3分),肾小球增殖性病变加重,间质浸润细胞减少。经过甲泼尼龙冲击及调整免疫抑制剂后缓解。2023年6月复查尿蛋白定量0.66 g/d,ANA滴度1∶512,SCr 72.5 μmol/L,补体C3 0.608 g/L,C4 0.084 4 g/L,当时的治疗方案为泼尼松10 mg联合吗替麦考酚酯2 g/d。
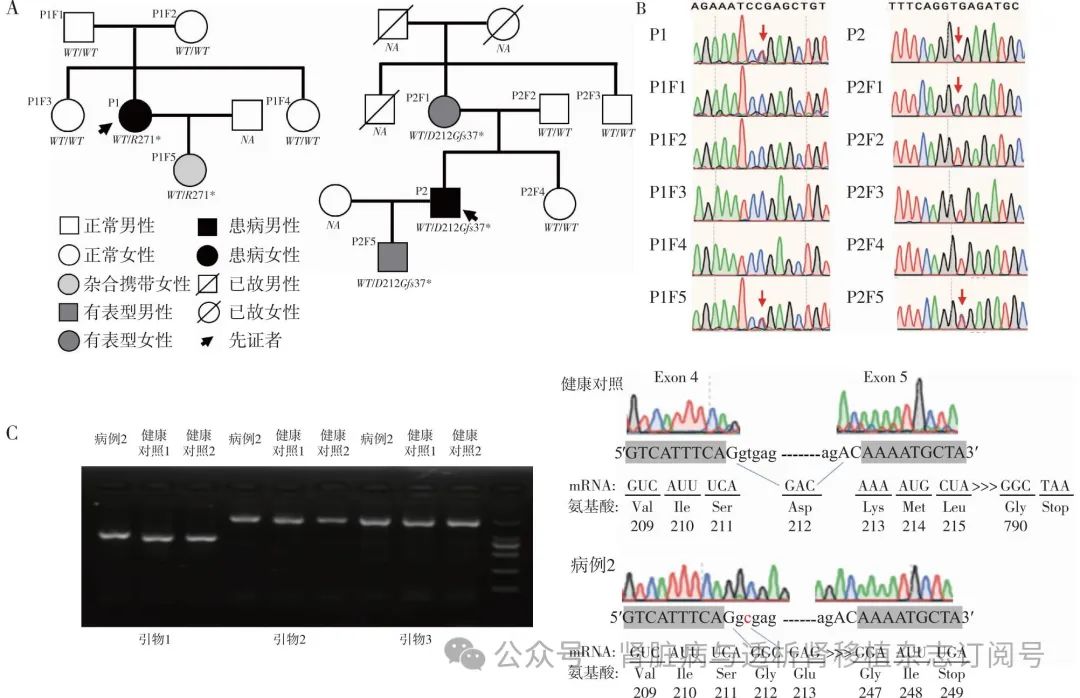
图1 家系图及剪切位点变异验证
P:患者;F:家系成员;R:精氨酸;D:天冬氨酸;G:甘氨酸;WT:野生型;A:2例患者的家系图及突变基因型;B:2例患者家系Sanger测序结果(红色箭头所示为突变位点);C:病例2 cDNA的Sanger测序图及胶图
病例2,男性,2015年8月(29岁)时出现双下肢轻度凹陷性水肿,伴前胸部和双上肢多发片状皮疹、脱发、口干和眼干,过敏原检测显示,对"霉菌"和"花粉"过敏,中药治疗无效。后出现肩关节疼痛,我院检查示尿蛋白定量7.29 g/d,尿沉渣红细胞计数16万/mL(多形型)。血清白蛋白17.8 g/L,SCr正常,血小板计数105×109/L,补体C3 0.167 g/L,C4 0.032 7 g/L,IgE>1 000.0 IU/mL,ANA滴度1:128,抗dsDNA阴性,抗核抗体谱SSA-Ro60、SSA-Ro52、核糖体P蛋白强阳性,其余阴性。胸部X线提示双侧胸腔积液。肾活检符合LN-Ⅳ型(AI 6分,CI 3分)。患者母亲有复发性无痛性口腔溃疡病史,其子被诊断为过敏性紫癜。
病例2经泼尼松联合吗替麦考酚酯治疗未见缓解,2015年12月加用他克莫司3 mg/d,4月后尿蛋白定量降至1.59 g/d。停用吗替麦考酚酯后,尿蛋白定量增加(7.04 g/d),C3轻度降低(0.688 g/L),加用雷公藤多苷后尿蛋白定量降至1.67 g/d,补体恢复正常。此后,泼尼松联合他克莫司维持治疗,持续少到中等量蛋白尿(0.38~1.28 g/d)。近期随访显示,患者肝肾功能正常,尿蛋白转阴,免疫学指标稳定,补体正常。
基因检测 通过全外显子组测序发现,病例1的6号染色体上TNFAIP3基因第6号外显子发生c.811C>T变异,使得第271位精氨酸被终止密码子取代(p.R271*)。Sanger测序对家系成员进行变异位点验证,发现其父母及两个姐妹未携带该突变,女儿携带但尚未出现临床症状(图1A、B)。该变异为无义突变,功能实验显示导致A20蛋白功能丧失(PVS1);经双亲验证为新发变异(PS2);千人基因组数据库、GnomAD数据库、ExAC数据库中正常人群中未发现该变异(PM2);生物信息学软件预测其影响有害(PP3);既往已在白塞病样自身炎症性疾病患者中检测到该变异(PP5),被ClinVar数据库收录为“致病性变异”。根据《美国医学遗传学和基因组学学会(ACMG)指南》,可以判断该变异为致病性变异。
病例2的TNFAIP3基因存在c.634+2T>C变异,为经典剪切位点变异,此前无相关报道,多种预测剪切的工具,包括regsnp_splicing, SpliceAI, Human Splicing Finder等预测该变异影响剪切。家系验证发现,患者的母亲及儿子携带该变异,父亲、妹妹及舅舅均不携带(图1A、B)。
为了进一步分析该变异对TNFAIP3 mRNA剪切的影响,我们提取了病例2的外周血RNA并合成cDNA进行Sanger测序,将其与健康对照进行比较,发现该变异导致TNFAIP3基因4号内含子保留,使得第37位氨基酸提前出现了终止密码子(p.D212Gfs37*)(图1C)。该变异影响TNFAIP3 mRNA剪切,导致移码突变(PVS1);数据库正常人群中未发现该变异(PM2);突变与疾病在家系中共分离(PP1)。根据《ACMG指南》,可以判断该变异为致病性变异。
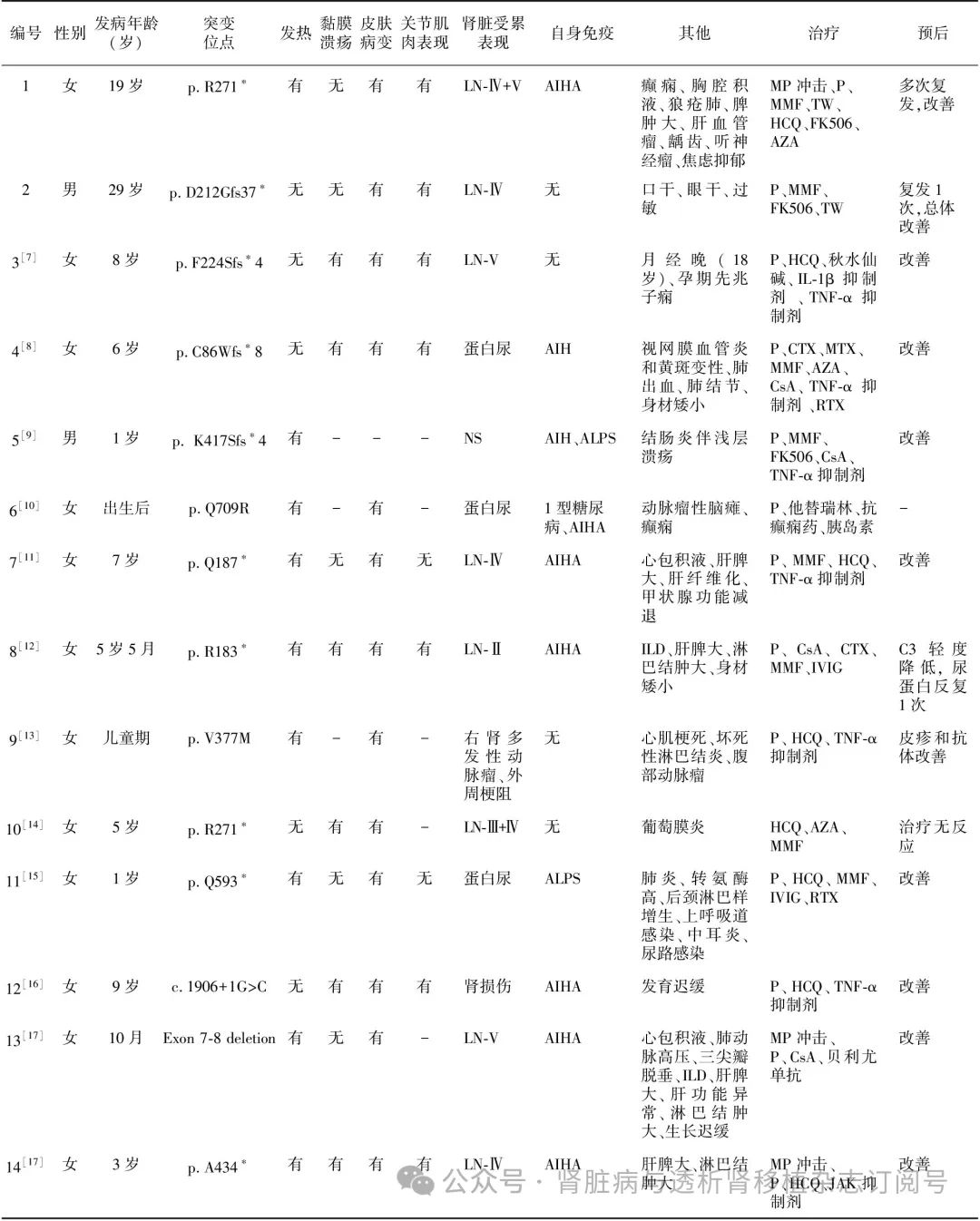
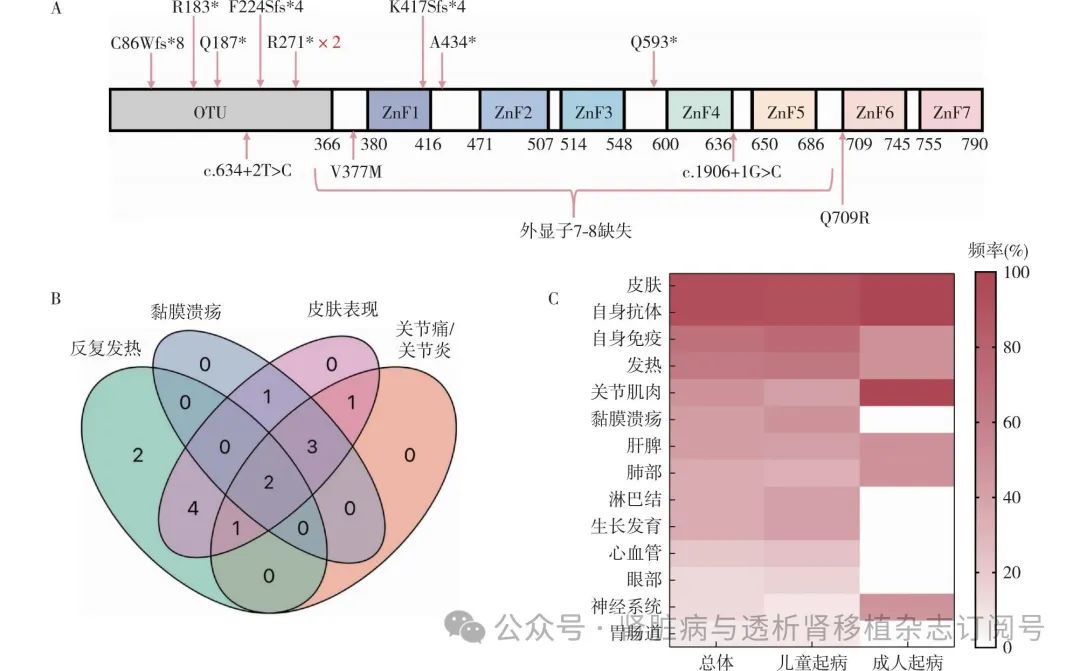
文献复习 经文献检索,全球已报道的近200例HA20患者中,21例确诊为SLE,除1例缺乏详细资料外,3例为成人期起病,17例为儿童期起病。21例SLE患者中14例伴有LN(含本文2例),且来自不同家系(表1)。共检测到13个TNFAIP3基因变异,均为杂合变异,8个无义或移码突变,2个剪切位点变异,1个拷贝数变异,2个错义突变(图2A)[6-17]。
14例患者中男性2例、女性12例,多儿童期起病,仅本文的2例成人期起病。92.86%的患者(13/14)有皮肤表现(皮疹、红斑、脱发等),64.29%(9/14)有发热,50%(7/14)有关节受累(关节炎、关节肿痛、关节积液等),42.86%(6/14)有黏膜溃疡(口腔、生殖器)(图2B)。13例(92.86%)患者自身抗体阳性,包括ANA,dsDNA,SSA、抗Sm抗体、抗核糖体P抗体、抗心磷脂抗体、类风湿因子等。10例(71.43%)患者合并多种自身免疫性疾病,包括自身免疫性溶血性贫血、自身免疫性肝炎、自身免疫性淋巴细胞增生综合征和1型糖尿病等。8例行肾活检,LN-Ⅳ型3例、LN-V型2例、LN-Ⅳ+V型、LN-Ⅲ+V型、LN-Ⅱ型各1例;6例未行肾活检,表现为肾病综合征1例,蛋白尿3例,右肾多发性动脉瘤1例。13例患者接受免疫抑制剂治疗,9例同时使用生物制剂[肿瘤坏死因子α(TNF-α)抑制剂6例,白细胞介素(IL)-β抑制剂1例,JAK抑制剂1例,利妥昔单抗2例,贝利尤单抗1例],11例症状和实验室指标得到改善,1例预后不详(表1)。
本文报道了2例表现为LN的TNFAIP3基因突变患者,分别存在TNFAIP3基因致病杂合变异R271*及D212Gfs37*,其中R271*变异为新发变异,D212Gfs37*变异遗传自母亲,并在该家系中呈现共分离。两例患者均为成年起病,此前相关报道中TNFAIP3变异患者大多幼年起病,成年发病较为少见。2例患者均出现了SLE典型的多器官受累及多种自身抗体阳性表现,较为特殊的是病例1严重的溶血性贫血与癫痫,以及病例2的过敏史。
与儿童期起病者相比,成年起病者黏膜溃疡、心血管、眼部、生长发育、胃肠道、淋巴结等病变风险较低,而关节、肌肉和神经系统受累比例更高(图2C)。但成人患者仅2例,需更多数据确认。幼年期的免疫系统尚未完全成熟,可能会引发更广泛的全身性炎症反应来应对免疫失调。因此,心血管、眼部、淋巴结等病变在幼年起病的患者中更显著,可能是由于这些系统在发育过程中对免疫异常的敏感度更高。
TNFAIP3基因位于6号染色体长臂,由TNF诱导,编码的A20蛋白包含N端的卵巢肿瘤区(OTU)结构域和C端的7个锌指区(ZnF)(图2A),广泛表达于多种免疫细胞。OTU域负责去泛素化,锌指结构调节泛素化,这种独特的结构使A20能够精细调控泛素依赖性信号转导。TNFAIP3基因突变遍布该基因各个区域,无热点突变,以蛋白截短突变为主,严重影响A20的功能。A20是NF-κB信号通路的重要负调控因子,抑制NF-κB激活,减少炎症因子的释放,并参与调控细胞存活与凋亡,维持细胞稳定,防止过度增生或凋亡[18]。
全基因组关联研究及病例对照研究发现多个TNFAIP3常见变异会增加SLE风险,随后的研究发现TNFAIP3罕见变异亦可导致SLE表现,提示常见变异与罕见变异可通过相同的生物学途径影响疾病的发生发展。我们既往在TNFAIP3基因突变患者外周血中观察到NF-κB通路及I型干扰素信号通路的过度激活[17],促炎因子TNF-α、IL-6的表达显著增加,导致组织损伤和炎症反应。TNF-α通过激活中性粒细胞、增强巨噬细胞和自然杀伤细胞的杀伤能力来刺激免疫系统抗感染,同时也是多种肾脏病病理过程中的关键介质,促进肾脏萎缩、肾小球内皮细胞损伤及纤维蛋白沉积[19]。IL-6是机体免疫反应的重要炎症因子,可导致肾脏内皮细胞功能障碍,增加血管通透性,促进炎症细胞和分子的渗透,加剧局部炎症反应[20]。过度的免疫反应还可能使免疫复合物在肾脏中沉积,激活补体系统并损伤肾小球滤过屏障,出现蛋白尿等肾脏表现。炎症因子过量表达导致肾脏组织受到过度的免疫攻击和炎症损伤,同时也加剧了自身免疫性疾病的风险,尤其是LN的发生。
表1 14例携带TNFAIP3变异LN患者的临床特征及预后
TNFAIP3:肿瘤坏死因子α诱导蛋白3;LN:狼疮性肾炎;AIH:自身免疫性肝炎;ALPS:自身免疫性淋巴细胞综合征;AIHA:自身免疫性溶血性贫血;NS:肾病综合征;ILD:间质性肺病;MP:甲泼尼龙;P:泼尼松;MMF:吗替麦考酚酯;TW:雷公藤多苷;FK506:他克莫司;AZA:硫唑嘌呤;HCQ:羟氯喹;CTX:环磷酰胺;MTX:甲氨蝶呤;CsA:环孢素A;IVIG:免疫球蛋白;IL-1β:白细胞介素1β;TNF-α:肿瘤坏死因子α;RTX:利妥昔单抗
TNFAIP3:肿瘤坏死因子α诱导蛋白3;LN:狼疮性肾炎;A:TNFAIP3编码的A20蛋白的结构图和14例LN患者TNFAIP3突变的位置;B:14例患者肾外表现情况;C:儿童/成人期起病的LN患者各系统累及对比
病例1病程中出现多次复发,但调整治疗后病情缓解。病例2经免疫抑制剂治疗,尿蛋白长期未完全缓解,但免疫学指标和肾功能正常。2例患者的治疗经历提示合理联用免疫抑制剂对TNFAIP3基因突变导致的LN治疗有效。文献回顾发现,IL-1β、TNF-α、JAK抑制剂可进一步改善此类患者的病情,显示了生物制剂的良好效果,但疗效需更多病例观察确认。
本研究存在以下局限性。首先,样本量有限,仅基于2例成人患者,关于成人期与儿童期发病临床差异的结论缺乏统计学支撑,需通过更大规模的研究进一步验证。其次,尽管本研究描述了免疫抑制剂和生物制剂的潜在效果,但缺乏对治疗策略的系统性评估和长期随访数据,限制了对疗效和预后的全面评估。因此,未来的研究应纳入更多病例,结合长期随访,进一步揭示TNFAIP3基因突变在LN中的致病机制,并优化治疗策略。
TNFAIP3基因变异谱广泛,其导致的LN临床表现缺乏特异性,常被误诊或漏诊,增加了诊断的难度。对合并多种自身免疫表现的LN患者,应详细询问家族史,及时完善基因检测,以帮助早期诊断和指导个体化治疗。

[本文引用]景小蔓, 张洋洋, 金英, 俞菁娴, 高行健, 张嘉会, 钟清, 刘志红, 张昌明. TNFAIP3基因突变导致成人狼疮性肾炎2例并文献复习[J]. 肾脏病与透析肾移植杂志, 2024, 33(5): 437-443.
JING Xiaoman, ZHANG Yangyang, JIN Ying, YU Jingxian, GAO Xingjian, ZHANG Jiahui, ZHONG Qing, LIU Zhihong, ZHANG Changming. TNFAIP3 gene mutations caused adult lupus nephritis in 2 cases and literature review[J]. Chinese Journal of Nephrology, Dialysis & Transplantation, 2024, 33(5): 437-443.
来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017