


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




丁文庆1 马洪闯2 综述 罗 群2 审校
DOI:10.3969/j.issn.1006-298X.2024.04.012
[基金项目] 浙江省宁波市共建医学重点学科-肾脏病学(2022-S03);浙江省公益技术应用研究计划项目(LGF22H050009)
[作者单位] 1宁波大学医学部(宁波,315211);2宁波市第二医院肾内科
摘 要
急性肾损伤(AKI)是临床常见的急危重症,发病率高、死亡率高。近年来研究表明,AKI后的肾脏适应不良性修复可导致慢性肾脏病(CKD)。铁死亡是一种新型的非凋亡形式的细胞死亡方式,其特征是铁过载和脂质过氧化,在AKI向CKD的进展中发挥重要作用。本文就铁死亡的经典调控机制及其在AKI进展为CKD中参与的病理机制作一综述。
关键词 急性肾损伤 慢性肾脏病 铁死亡 炎症 纤维化
Ferroptosis in the progression from acute kidney injury to chronic kidney disease
DING Wenqing1,MA Hongchuang2,LUO Qun2
1Health Science Center,Ningbo University,Ningbo 315211,China
2Department of Nephrology,Ningbo No.2 Hospital,Ningbo 315010,China
ABSTRACT
Acute kidney injury (AKI) is an acute and critical illness with high morbidity and mortality.In recent years,a growing body of studies have shown that renal maladaptation following AKI results in chronic kidney disease (CKD).Ferroptosis,a novel form of non-apoptotic cell death characterized by iron overload and lipid peroxidation,is believed to play a significant role in the progression of AKI to CKD.This article provides a review of the classical regulatory mechanisms of ferroptosis and its potential involvement in the pathological mechanisms in the progression of AKI to CKD.
Key words acute kidney injury chronic kidney disease ferroptosis inflammation fibrosis
急性肾损伤(AKI)患者肾功能快速下降,是临床常见的急危重症,发病率高、死亡率高,其发生机制复杂,主要包括肾脏缺血再灌注损伤、肾毒性物质引起的肾小管细胞损伤、炎症反应和免疫介导的肾脏损害[1]。研究表明,AKI是慢性肾脏病(CKD)的独立危险因素,AKI后的肾脏适应不良性修复可导致CKD进展[2]。这种适应不良性修复涉及炎症、缺氧、间质纤维化及肾小管上皮细胞损伤等病理机制[3-4]。铁死亡与AKI的发病机制密切相关,且其发生促进AKI向CKD的转变,因此靶向铁死亡具有良好的治疗前景。本文综述了铁死亡的经典调控机制及其在AKI进展为CKD中参与的病理机制。
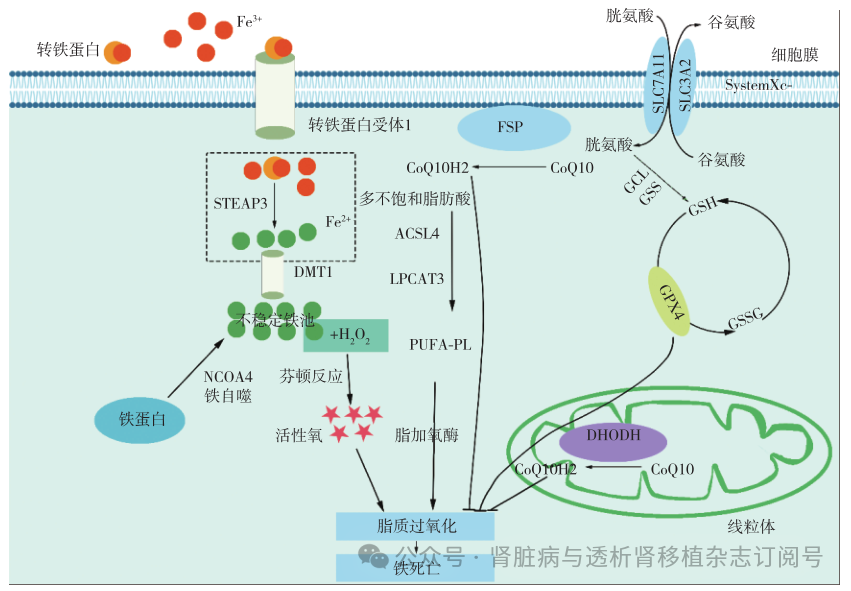
铁死亡由Dixon等[5]在2012年首次命名,是一种铁依赖的磷脂过氧化作用驱动的细胞死亡模式,具有独特的形态学和生化特征。形态学上,铁死亡会导致细胞线粒体嵴减少、消失,线粒体膜破裂、皱缩;生化上,铁死亡是一种依赖活性氧(ROS)的细胞死亡形式,与铁过载和脂质过氧化相关[6]。铁死亡受到多种细胞内信号途径调控(图1)[5,7]。
图1 铁死亡的主要信号通路和关键调节因子[5,7]
STEAP3:前列腺六跨膜上皮抗原3;DMT1:二价金属离子转运体1;FSP:铁死亡抑制蛋白;CoQ10:辅酶Q10;CoQ10H2:还原型辅酶Q10;ACSL4:酰基辅酶A合成酶长链家族成员4;LPCAT3:溶血磷脂酰胆碱酰基转移酶3;PUFA-PLs:多不饱和脂肪酸磷脂;System Xc-:胱氨酸/谷氨酸反向转运体;GCL:谷氨酸-半胱氨酸连接酶;GSS:谷胱甘肽合成酶;GSH:谷胱甘肽;GPX4:谷胱甘肽过氧化物酶 4;DHODH:二氢乳清酸脱氢酶;NCOA4:核受体共激活剂4
胱氨酸/谷氨酸反向转运体(System Xc-)/过氧化物酶4(GPX4)途径 System Xc-是细胞膜上的跨膜转运复合物,由轻链亚基(SLC7A11)和重链亚基构成,介导谷氨酸和胱氨酸的等比例交换[8]。胱氨酸还原产生的半胱氨酸是合成抗氧化剂谷胱甘肽(GSH)的限速底物[9],因此抑制System Xc-可减少GSH表达,促进氧化损伤,最终导致铁死亡。GPX4是哺乳动物细胞中还原脂质过氧化产物的主要酶,GSH作为其辅因子,协助GPX4清除膜脂质过氧化物[9]。研究表明,P53通过抑制SLC7A11的表达来降低System Xc-的活性,减少胱氨酸的摄入,改变GPX4的活性来促进铁死亡[10]。
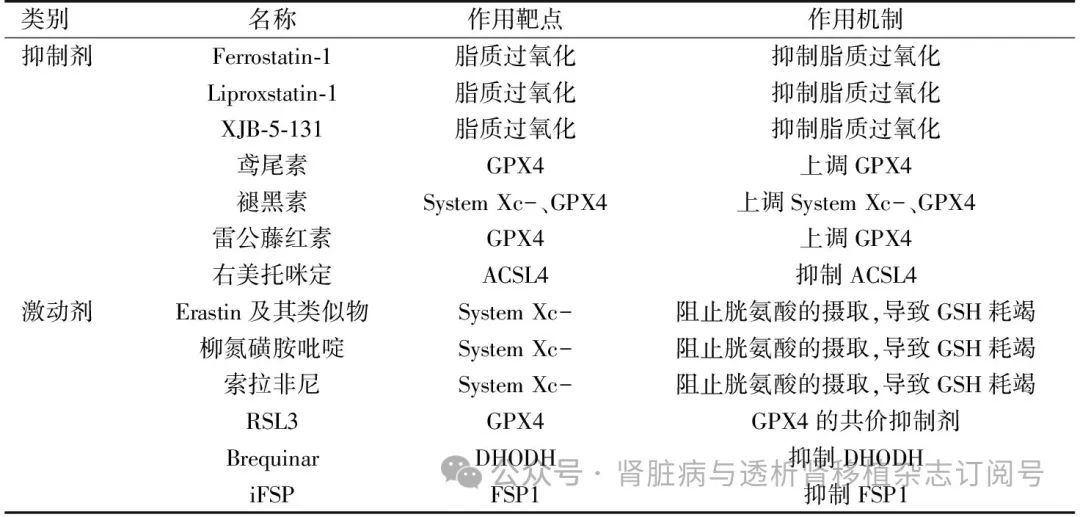
表1 铁死亡调节剂
GPX4:谷胱甘肽过氧化物酶4;System Xc-:胱氨酸/谷氨酸反向转运体;ACSL4:酰基辅酶A合成酶长链家族成员4;GSH:谷胱甘肽;DHODH:二氢乳清酸脱氢酶:FSP:铁死亡抑制蛋白;RSL3:谷胱甘肽过氧化物酶4抑制剂
铁代谢途径 铁过载是铁死亡的典型特征。Fe3+与转铁蛋白结合,通过转铁蛋白受体1进入细胞,随后被还原为Fe2+并储存在不稳定铁池(LIP)中。病理状态下,过量Fe2+通过芬顿反应增加ROS生成,引发铁死亡[6]。铁蛋白由铁蛋白重链(FTH)和铁蛋白轻链组成,FTH具有亚铁氧化酶活性,可将铁维持在无毒的Fe3+形式,防止芬顿反应及其引起的氧化损伤[11]。FTH基因缺失会增加ROS产生,引发铁死亡[7]。核受体共激活剂4(NCOA4)通过促进铁蛋白降解诱导铁过载,推动铁死亡[11]。
脂质代谢途径 脂质过氧化是铁死亡的另一典型特征。多不饱和脂肪酸(PUFA)易发生脂质过氧化,其中含有花生四烯酸(AA)的磷脂酰乙醇胺是铁死亡过程中主要的氧化底物[12]。游离PUFA在酰基辅酶A合成酶长链家族成员4(ACSL4)和溶血磷脂酰胆碱酰基转移酶3作用下生成细胞膜磷脂,随后在脂加氧酶(LOX)的催化下生成脂质过氧化物,破坏细胞膜,引发铁死亡[6]。ACSL4是铁死亡的已知启动子,敲除ACSL4可抑制铁死亡[13],这揭示了通过阻断含PUFA的膜磷脂生物合成途径可有效抑制铁死亡。
GPX4非依赖性铁死亡途径 除了经典的System Xc-/GPX4轴外,最近的研究发现了几种GPX4非依赖性铁死亡调控机制,包括铁死亡抑制蛋白 1(FSP1)/辅酶Q10(CoQ10)途径和二氢乳清酸脱氢酶(DHODH)/CoQ10途径。FSP1位于细胞膜上,具有泛醌氧化还原酶活性,能够将CoQ10还原为抗氧化的还原型CoQ10(CoQ10H2),抑制脂质过氧化物形成[12]。DHODH位于线粒体内膜,在嘧啶合成中将二氢乳清酸(DHO)氧化为乳清酸(OA),同时将CoQ10还原为CoQ10H2[14]。FSP1/CoQ10轴和DHODH/CoQ10轴协同维持高水平的CoQ10H2,共同增强细胞抗氧化能力,提高对铁死亡的抵抗力。
炎症 慢性炎症是推动AKI向CKD进展的关键因素[4]。PUFA在炎症中起重要作用。AA被环氧化酶(COX)和LOX代谢成前列腺素和白三烯、脂氧素等炎性介质。虽然前列腺素内过氧化物合酶2(PTGS2/COX2) 通常作为铁死亡的生物标志物,但在某些炎症条件下的上调并不总与铁死亡相关[15]。在铁死亡过程中,损伤相关分子模式(DAMP)的释放能触发固有免疫系统。DAMP通过与模式识别受体结合引发炎症反应。高迁移率族蛋白B1(HMGB1)是坏死细胞释放的DAMP。HMGB1中和抗体能减轻铁死亡诱导的巨噬细胞炎症反应,表明HMGB1可能是抑制与铁死亡相关炎症的潜在治疗靶点。Zhao等[16]研究进一步揭示了HMGB1的双重作用:不仅作为DAMP参与AKI,还通过增加肾小管细胞对持续氧化应激的敏感度,延缓AKI向CKD的转变。这些研究支持了DAMP在组织炎症和铁死亡调节中的作用。ACSL是脂质过氧化过程中的重要酶,也参与炎症反应。Wang等[13]发现敲除ACSL4显著减少AKI小鼠的铁死亡及肾脏巨噬细胞的浸润。Tao等[17]同样发现右美托咪定通过α2肾上腺素受体抑制ACSL4,减轻铁死亡介导的肾缺血再灌注损伤和炎症。铁死亡还可能触发炎症级联反应。研究表明,抑制肾小管上皮细胞的铁死亡能通过降低单核细胞趋化蛋白1(MCP-1)的分泌来减少巨噬细胞的招募,减轻炎症反应[18]。此外,铁死亡还能通过招募巨噬细胞间接促进中性粒细胞的聚集[13],为铁死亡在AKI中的促炎作用提供了另一种机制。铁死亡抑制剂在AKI中显示出抗炎作用,例如鸢尾素通过上调GPX4来下调炎症反应并抑制铁死亡[19]。以上实验表明,铁死亡不仅可能触发炎症反应,而且可能在一定程度上加剧了炎症的发展。
缺氧 缺氧是AKI向CKD转化的关键病理生理机制[3]。AKI后的毛细血管稀疏和内皮细胞受损会导致肾缺氧,诱发肾小管间质纤维化,加速AKI向CKD转变[4]。研究表明,多种转录因子在缺氧环境中能保护肾脏细胞免受损伤,并通过调控铁死亡相关病理状态发挥作用。核因子E2相关因子2(Nrf2)是抗氧化防御系统的关键调节因子,调控多种抗氧化酶的表达[20]。褪黑素能通过激活Nrf2在缺氧/再灌注氧小鼠肾小管上皮细胞中抑制氧化应激并阻止铁死亡,从而预防AKI的发生[21]。缺氧诱导因子(HIF)由HIF-α和HIF-β亚基组成。在缺氧条件下,细胞会上调HIF以适应低氧环境[20]。Li等[22]利用叶酸诱导的肾损伤模型证实,通过HIF脯氨酰羟化酶抑制剂-罗沙司他预处理可稳定HIF-1α和Nrf2信号通路,同时减少铁死亡,抑制炎症,并延缓肾纤维化的进程。HIF-1α的下调可能会导致ACSL4的过度表达,促进铁死亡[13]。另有研究指出,Nrf2介导缺氧诱导的HIF-1α激活,Nrf2和HIF-1α相互作用,在减轻缺血性AKI方面起重要作用[20]。阻遏因子1沉默转录因子(REST)是一种缺氧时基因表达抑制的主要调节因子,在各种AKI模型中表达上调[23]。REST通过抑制谷氨酸胱氨酸连接酶修饰亚基(GCLM)的转录,减少GSH和GPX4的水平,促进脂质过氧化和铁死亡,最终诱导AKI及其向CKD的进展。特异性敲除肾小管上皮细胞的REST基因能显著改善上述效应[23]。研究显示,在缺氧条件下的AKI,损伤会通过一种称为同步铁死亡的机制在小鼠近端肾小管上皮细胞中传播,引发相邻细胞的连续死亡[24]。Wang等[25]发现,在缺氧条件下,小细胞外囊泡在近端肾小管上皮细胞顶端膜分泌增加,并诱导近端肾小管上皮细胞发生铁死亡,铁死亡抑制剂和RNA酶A能有效抑制这一过程。这表明小细胞外囊泡在铁死亡信号传递中起关键作用。这些研究揭示了Nrf2、HIF及REST等转录因子在调控铁死亡过程中对AKI向CKD转变的关键作用,为开发新的治疗靶点提供了方向。
肾间质纤维化 肾间质纤维化是肾脏适应不良性修复过程中的一个标志,是多种病因所致CKD的共同终点。在多种AKI模型中观察到铁死亡参与纤维化的发生[18,22]。肌成纤维细胞激活和大量细胞外基质分泌是间质纤维化的核心特征。转化生长因子β(TGF-β)/Smad是肾纤维化主要的信号通路,且与铁死亡紧密相关。Chen等[26]研究发现组蛋白去乙酰化酶3(HDAC3)在促进纤维化过程中起重要作用,其表达受到TGF-β/Smad信号通路的调控,通过使用HDAC3选择性抑制剂能够减轻对抗纤维蛋白Klotho的抑制,发挥抗肾纤维化作用。在马兜铃酸和叶酸诱导的AKI向CKD转变小鼠模型中,Zhang等[27]观察到HDAC3异常升高,并伴随GPX4表达下调,导致肾小管上皮细胞发生铁死亡。特异性敲除HDAC3后,GPX4表达上调,肾纤维化显著改善。这些发现表明通过靶向HDAC3可延缓AKI向CKD的进展。上皮间质转变(EMT)是纤维化过程中另一关键环节。研究表明,在EMT过程中可见FTH减少,铁蛋白释放游离铁离子引发铁过载,导致ROS产生过剩,触发铁死亡[18]。在EMT过程中减少外源性FTH,纤维化程度加重[18]。这表明铁稳态对预防EMT和肾脏纤维化进展的重要性。健康的肾小管上皮细胞主要依赖脂肪酸氧化(FAO)作为能量来源,肾损伤后,肾小管上皮细胞的FAO能力受损[28],研究表明,低水平的FAO也可能导致肾脏纤维化[29]。通过靶向铁死亡能有效减轻肾间质纤维化的程度,改善肾功能,延缓AKI向CKD的进展。
肾小管上皮细胞损伤 肾小管上皮细胞损伤不仅是AKI的典型表现,还推动AKI向CKD进展。近端小管作为物质转运和重吸收的关键部位,对能量的需求巨大。肾小管上皮细胞内富含线粒体,通过FAO产生所需的ATP[29]。AKI后,肾小管受损导致线粒体功能障碍,线粒体活性氧积累,触发铁死亡[30]。受损的肾小管上皮细胞通过多种机制促进炎症和纤维化,例如转变为分泌表型并释放促炎因子和促纤维化因子[31]。AKI后如果损伤持续存在,肾小管上皮细胞无法完成正常修复,发生G2/M期阻滞,进而通过旁分泌机制在纤维化过程中发挥作用[31]。靶向肾小管上皮细胞铁死亡可有效缓解炎症和纤维化,延缓CKD进展。XJB-5-131是一种针对线粒体的硝基氧化物,具有双重抗氧化效应,能通过抑制脂质过氧化阻止铁死亡。研究发现,XJB-5-131通过特异性抑制铁死亡,减少炎症细胞浸润和促炎细胞因子表达,促进肾小管上皮细胞增殖和修复[32]。通过靶向特定基因也可显著改善AKI预后。例如,在近端小管中敲除FTH基因后,AKI小鼠的存活率显著下降[11]。而特异性敲除肾小管上皮细胞中的ACSL4则能显著减少炎症和巨噬细胞浸润,减轻脂质过氧化,预防铁死亡介导的AKI[13]。
铁死亡调节剂干预AKI向CKD进展
铁死亡调节剂通过靶向铁死亡的不同靶点(表1),预防AKI进展为CKD,从而发挥肾脏保护作用。抑制铁死亡可减轻急性肾组织损伤后的炎症,这可能跟DAMP的释放减少相关。铁死亡抑制剂Ferrostatin-1可阻止白细胞介素33 (IL-33)的加工和释放,抑制巨噬细胞浸润,避免进一步的肾损伤[33]。在缺氧环境下,雷公藤红素通过激活Nrf2介导GPX4上调,抑制铁死亡,在顺铂诱导的AKI治疗中具有重要作用[34]。一些铁死亡抑制剂还能改善肾间质纤维化,Liproxstatin-1通过减少人肾皮质近曲小管上皮细胞在铁死亡期间分泌的促纤维因子,抑制成纤维细胞活化,减缓纤维化进程[35]。此外,罗沙司他、鸢尾素均被证明能够抑制纤维化的进展[19,22]。目前这些药物的研究仍限于实验阶段,未来需进一步深入探索,为临床应用奠定基础。
小结:本文综述了铁死亡的主要信号通路及其在AKI向CKD进展中的作用。将铁死亡作为治疗靶点有望延缓AKI向CKD的进程,且具有良好的应用前景。随着基因编辑和纳米技术的发展,精准诱导铁死亡成为可能,有望提高疗效。然而,铁死亡的调控仍面临非靶向细胞死亡、铁死亡机制未明、动物模型向临床转化困难等挑战。未来的研究需要深入探索AKI、CKD和铁死亡之间的相互作用机制,通过开发精准的靶向治疗策略,降低AKI的发病率,延缓CKD进展,改善患者预后。
参考文献


来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017