


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




炎性肌病是一组以肌肉炎症和无力为特征的自身免疫性疾病,给患者带来严重的致残后果和生活质量下降。近年来,随着对这类疾病认识的深入,研究者们发现线粒体功能障碍在某些炎性肌病中扮演着重要角色,尤其是在包涵体肌炎(IBM)中,线粒体异常已被公认为其特征性病理改变之一。然而,对于非IBM型炎性肌病中线粒体病理的临床意义,目前学界尚缺乏全面深入的研究。
最近发表在Journal of Neurology上的一项多中心研究对这一问题进行了探讨。该研究来自欧洲六个神经肌肉中心,包括意大利、法国和德国的研究团队,他们对850名炎性肌病患者的肌肉活检标本进行了回顾性分析。研究旨在确定非IBM型炎性肌病中线粒体病理的发生率及其临床意义,评估线粒体异常是否可作为疾病向IBM进展、治疗反应和临床预后的潜在标志物。
研究团队按严格标准纳入了25例非IBM型炎性肌病患者,这些患者的肌肉活检显示了超出年龄预期的细胞色素C氧化酶(COX)阴性肌纤维比例。对于40岁以下患者,COX阴性纤维超过0.25%即被视为病理性;40至60岁患者超过0.5%;60岁以上患者超过1%。研究者还选取了20名没有明显线粒体异常的炎性肌病患者作为对照组。
这25名患者中,女性占68%,平均发病年龄为58.8岁。在肌病亚型分布上,伴有线粒体病理的多肌炎(PM-Mito)和非特异性肌炎(NSM)最为常见,共占72%。肌肉活检中COX阴性纤维的平均比例为3%(范围0.25-8.5%),这些纤维的分布模式在不同肌病亚型间有所差异。免疫介导的坏死性肌病(IMNM)、抗合成酶综合征(ASyS)和重叠性肌炎(OM)中呈散在分布,而皮肌炎(DM)样本则显示弥漫性分布。PM-Mito和NSM的分布模式更为多样,NSM倾向于散在分布(66%),而PM-Mito更常呈弥漫性分布(77%)。
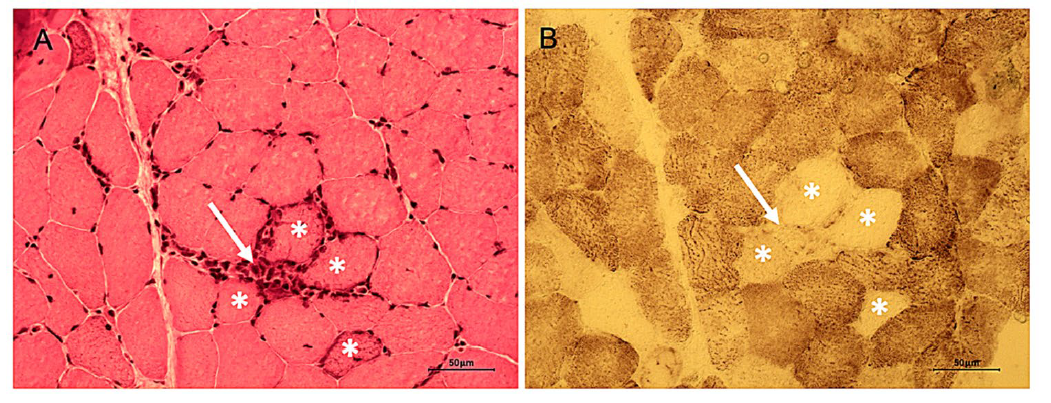
图:线粒体改变与炎症在一例非特异性肌炎(NSM)患者中的关系。A:苏木精-伊红(H&E)染色显示炎性细胞浸润(黄色箭头)主要分布在肌内膜,围绕着成群的肌纤维(星号)。B:对相同区域进行细胞色素c氧化酶(COX)染色,显示出COX缺陷的肌纤维(星号),提示该区域存在线粒体功能障碍,这可能是继发于炎症所致。比例尺 = 50 μm
炎症浸润在13例(52%)患者中以内膜为主,5例(20%)以周膜为主,7例(28%)兼有两处浸润。大部分患者(72%)的炎症浸润呈簇状,其余(28%)则为散在的孤立细胞。值得注意的是,在三例患者中,研究者观察到COX阴性纤维的分布与炎症浸润部位密切相关,这提示局部炎症过程可能直接导致线粒体功能障碍。然而,在无炎症浸润区域也能观察到散在的COX阴性纤维,表明可能存在其他或额外的致病机制。
所有患者的肌肉活检均显示主要组织相容性复合物I(MHC-I)的过度表达,这是炎性肌病的典型特征。在临床表现方面,8例(32%)患者表现为全身性无力,17例(68%)以近端无力为主,14例(56%)伴有轴性无力。血清肌酸激酶(CK)平均值为3065 UI/L(范围189-14000)。
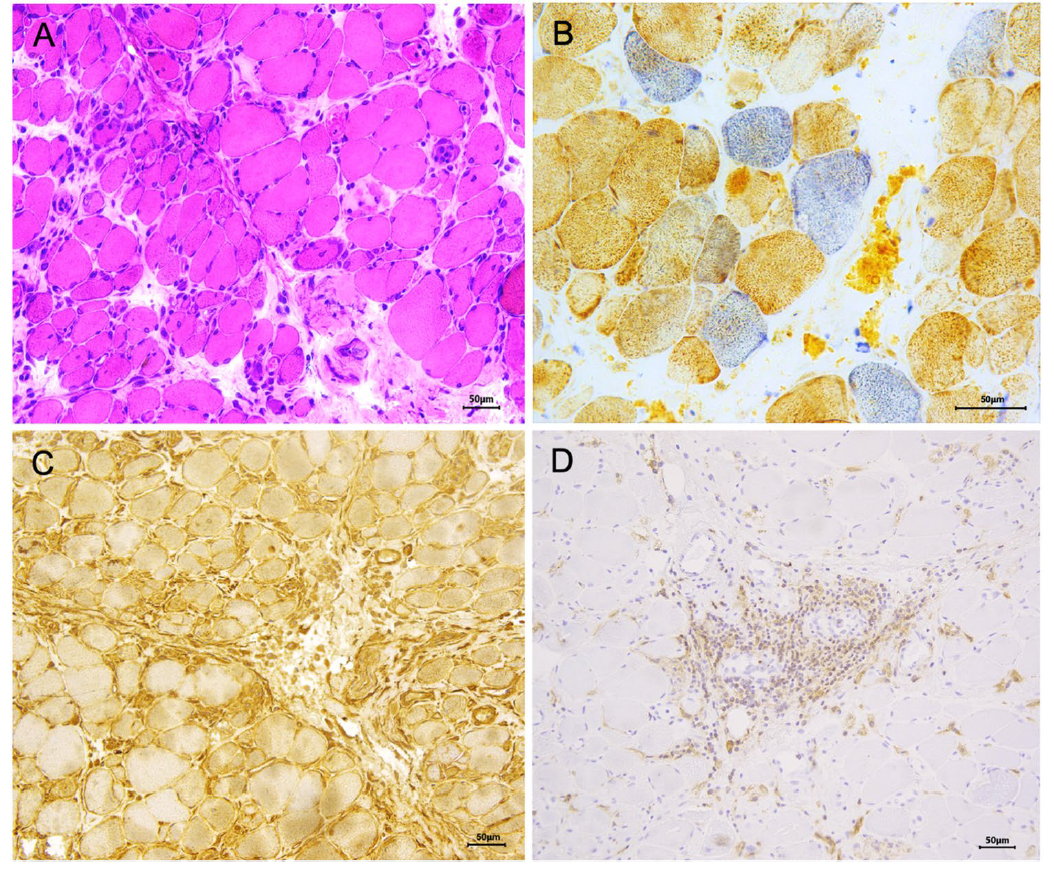
图:代表性肌肉活检显示抗Mi-2皮肌炎存在弥漫性线粒体改变。A:肌纤维坏死及再生,并伴有内肌间隙和肌束间隙(HE)炎症细胞浸润;B:通过COX/SDH染色可见大量COX阴性的肌纤维,提示明显的线粒体功能障碍;C:MHC-I在肌质和肌膜区域弥漫性高表达;D:可见显著T细胞淋巴细胞浸润(CD4)。缩写注释:CD4,分化簇4;COX,细胞色素c氧化酶;HE,苏木精-伊红染色;MHC-I,I型主要组织相容性复合体;SDH,琥珀酸脱氢酶
研究中最引人关注的发现是,线粒体病理与治疗抵抗和不良临床预后显著相关。与对照组相比,伴有线粒体异常的患者临床预后明显较差(p=0.003)。此外,COX阴性纤维比例越高,临床预后越差(p=0.031),研究者发现当COX阴性纤维比例达到2.5%以上时,患者临床预后评分多为2分或更高(代表中重度持续性肌无力)。这一结果强烈提示线粒体功能障碍可能是非IBM型炎性肌病严重程度和治疗无效性的重要标志物。
更为重要的是,研究过程中发现4名患者(2名PM-Mito和2名NSM)在疾病过程中进展为临床确诊的IBM。与其他患者相比,这些患者的COX阴性纤维比例明显更高(平均3.5%对比其他患者,p=0.029),这支持线粒体功能障碍程度可能与向IBM进展的风险相关的假说。这四名患者均表现出对治疗的抵抗性和较差的预后,最终导致中重度残余肌无力,其中两例需要使用轮椅。
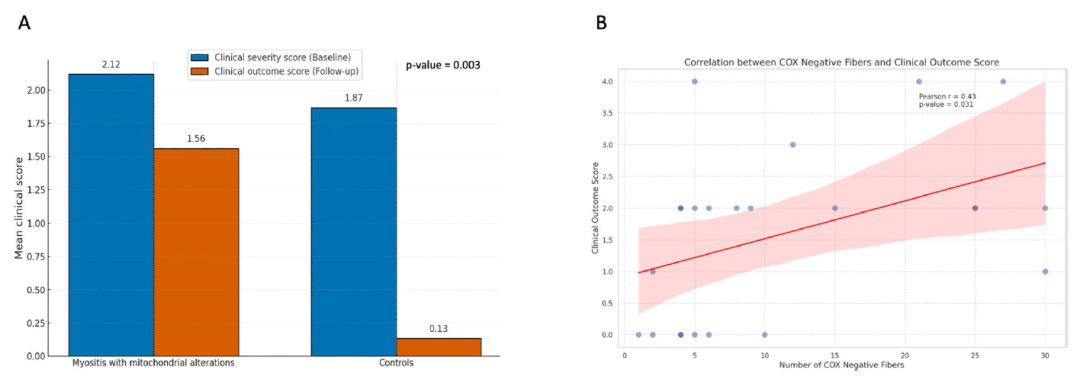
图:肌肉线粒体异常与较差临床结局之间的相关性。A 图表显示了在症状出现时(基线)和随访时,对于伴有线粒体异常的肌炎患者与无线粒体异常的对照组患者的临床严重程度评分和临床结局评分的比较。评分越高分别表示疾病越严重及临床结局越差。线粒体异常肌炎组患者在两个时间点的临床评分均明显高于对照组,随访时平均临床结局评分为1.56,而对照组平均得分仅为0.13(p = 0.003)。这一显著差异表明,肌炎患者出现线粒体异常与持续的临床功能障碍相关,而无相关线粒体异常的对照组则无此表现。临床评分分级如下:0 = 无症状(无临床功能障碍),1 = 轻度症状伴轻微肌无力(MRC ≥ 4);2 = 中到重度肌无力(MRC < 4),但仍能行走;3 = 严重肌无力,需用一根或两根拐杖辅助行走;4 = 严重肌无力,需要轮椅或卧床。B 散点图以回归线显示了伴有线粒体异常的肌炎患者中COX阴性肌纤维数量与临床结局评分之间的关系。X轴表示肌肉活检样本中检测到的COX阴性纤维数,数量越多表示线粒体功能障碍越严重。Y轴表示临床结局评分,数值越高说明患者的临床结局越差。图中显示COX阴性纤维数量与临床结局评分之间存在正相关关系,提示COX阴性纤维数量较多的患者临床结局较差(p = 0.031)。
研究者们还对线粒体病理的分布模式与疾病进展的关系进行了探讨。尽管未能确定能可靠区分将来会进展为IBM的病例的特定分布模式,但数据显示,后来进展为IBM的PM-Mito和NSM患者通常具有更高的COX阴性纤维负担,支持更广泛的线粒体功能障碍可能与更差的临床轨迹相关。
值得一提的是,研究还发现非IBM型炎性肌病患者从症状出现到诊断的时间间隔越长,肌肉中的线粒体异常就越显著(p=0.0298)。这可能是由于早期症状较轻微或非典型临床表现,导致诊断延迟。此外,线粒体功能障碍可能是逐渐发展的过程,使早期识别变得困难。
PM-Mito作为一种特殊的肌炎亚型,与多肌炎和IBM共享一些特征。它具有多肌炎的组织学特点和IBM的临床特点,如股四头肌或手指屈肌的受累,但肌纤维中没有特征性的边缘空泡。研究者提出,PM-Mito可能是IBM的早期阶段,或者是一种倾向于随时间进展为IBM的独特实体。本研究中观察到的从非特异性肌炎到IBM的潜在进展路径似乎始于早期的、伴有显著线粒体病理的非特异性肌炎,其中特征性症状如股四头肌或手指屈肌无力尚未出现。这一早期阶段可能演变为伴有线粒体受累的多肌炎(PM-Mito),最终导致包涵体肌炎(IBM)。这一可能的疾病谱系强调了在早期阶段诊断IBM的困难,因为初始症状往往微妙且非特异。随着疾病的进展,IBM的临床表现会变得更加明显。重要的是,尽管从非特异性肌炎到IBM的过渡可能需要相当长的时间(>10年),但从PM-Mito到IBM的进展可能发生得更快。
关于线粒体功能障碍在炎性肌病中的病理机制,研究者指出目前尚存争议。一些研究表明,在皮肌炎和多肌炎中,氧化代谢缺陷(线粒体功能障碍)可能是血液供应受损的继发结果。而另一方面,线粒体功能障碍会增加活性氧(ROS)的产生,驱动干扰素(IFN)诱导基因的表达和肌肉炎症,这反过来又会导致线粒体功能异常,形成疾病的自我维持循环。
最近的研究发现,位于线粒体的细胞凋亡激活因子harakiri(HRK)在肌炎骨骼肌细胞中上调。HRK表达升高导致线粒体电位降低和损伤修复能力下降。Toll样受体7(TLR7)激活并维持肌炎患者细胞中高水平的HRK表达,导致线粒体损伤,这可能与卫星细胞调节障碍一起,导致肌纤维修复受损、肌纤维变性和肌肉无力。因此,本研究的数据表明,肌炎中的线粒体功能障碍可能对肌肉组织的修复和再生能力产生负面影响,导致治疗抵抗性。
这项研究也存在一些局限性。首先,作为回顾性分析,可能存在选择偏倚,且相对较小的队列规模(25名患者)限制了研究结果的普遍适用性。其次,缺乏纵向肌肉活检数据,尤其是对那些进展为IBM的患者,这使得难以详细了解病理变化的演变过程。此外,缺乏影像学和血清学数据,如抗cN1A抗体状态,也限制了对疾病进展的全面评估。最后,缺乏电子显微镜和遗传学数据,限制了对线粒体功能障碍更深层次的超微结构和遗传贡献因素的探索。
尽管如此,这项研究的结果仍具有重要的临床意义。线粒体病理在肌炎患者中的存在与更差的临床预后和治疗抵抗性显著相关,更高比例的COX阴性肌纤维也与更差的临床预后相关。COX阴性肌纤维的存在可能预示随时间进展为IBM的风险增加,因此建议对这些患者进行更严密的临床监测,以避免在IBM表现明显时进行不必要的治疗。肌肉活检中的线粒体病理代表非IBM型炎性肌病疾病严重程度和治疗无效性的潜在生物标志物。
总之,这项多中心研究为理解线粒体功能障碍在非IBM型炎性肌病中的临床意义提供了重要见解。研究结果表明,线粒体病理不仅是IBM的特征,也可能在其他炎性肌病中发挥重要作用,尤其是与预后和治疗反应相关。

来源:神经科的那些事
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017