


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




空军军医大学西京医院呼吸与危重症医学科 杨丽娟 宋立强
44岁男性患者,发现双上肺阴影10年,咳嗽、活动后气短2年余。
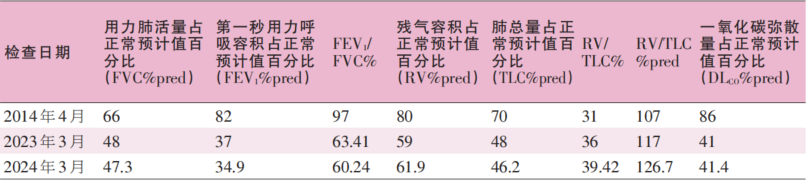
2014年4月,患者体检查胸部计算机体层摄影(CT)示双肺散在斑片状密度增高影及小腺泡影,边界模糊,双上肺为著,查血常规、结核抗体、肿瘤系列、自身抗体、痰液相关检查均阴性,经支气管镜活检提示支气管黏膜、肺组织慢性炎伴炎性渗出及间质纤维组织增生,肺功能提示轻度限制性损害,弥散功能正常(表1)。予以甲磺酸左氧氟沙星治疗,后复查胸部CT较用药前无明显变化,因无其他特殊不适,未予重视。
2021年9月,患者受凉后出现咳嗽、咳白痰、活动后气短,不伴发热,胸部CT示双肺间质纤维化伴感染,双肺上叶肺大疱,双肺支气管牵拉扩张,结核感染T细胞斑点试验(T-SPOT)阴性。予以甘氨酸茶碱钠缓释片、沙丁胺醇气雾剂、阿莫西林克拉维酸钾片治疗,用药后症状较前稍好转,但仍有间断性咳嗽、活动后气短,未作进一步处理。
2023年3月,患者受凉后咳嗽,气短加重,伴发热,体温最高39℃,入住当地结核病院,查胸部CT示双肺弥漫性病变伴两侧中上野毁损、囊变、支气管牵拉扩张,双侧肺门上提,双侧胸膜增厚、粘连,双侧胸腔少量积液,血常规示白细胞计数(WBC)14.05×109/L,中性粒细胞百分数(NEUT%)90.70%,红细胞沉降率(ESR)72 mm/h,新型冠状病毒核酸阴性,甲流/乙流抗原阴性,T-SPOT检查阴性,痰培养分枝杆菌阴性,肺功能提示重度混合性通气功能障碍(表1)。诊断为“1.细菌性肺炎;2.陈旧性肺结核;3.毁损肺”。予以抗感染、对症治疗后症状稍好转,复查胸部CT示右肺病变较前略有吸收,其余病灶未见变化,于4月9日出院,出院后遵医嘱开始异烟肼0.3 g qd po+利福喷丁0.6g biw po抗结核治疗。患者抗结核治疗期间每月复查一次胸部CT,2024年3月复查胸部CT较前无明显变化,且咳嗽,活动后气短症状逐渐加重,遂于2024年3月8日就诊于我科。
表1 患者不同时期肺功能变化情况
患者无高血压、糖尿病、冠心病、慢性阻塞性肺疾病等慢性疾病史,无药物、毒物摄入史。吸烟10余年,每天约20支,已戒烟4年。婚育史、家族史无特殊。
体温36.7℃,脉搏72次/分,呼吸22次/分,血压98/70 mmHg。患者体形消瘦,体质指数(BMI)18.5 kg/m2。全身浅表淋巴结未触及肿大。双肺呼吸音粗,可闻及干啰音。心脏及腹部查体未见异常。双下肢无浮肿。无杵状指(趾)。
pH值为7.379,动脉血氧分压(PaO2)为80.9 mmHg,动脉血二氧化碳分压(PaCO2)为43.3 mmHg,血清碳酸氢盐(HCO3-)为25.5 mmol/L,动脉血氧饱和度(SaO2)为97%。
WBC 4.56×109/L,血红蛋白(Hb)147 g/L,血小板计数(PLT)169×109/L,NEUT%69.7%,淋巴细胞百分数(LYMPH%)19%,嗜酸性粒细胞百分数(EO%)4.1%。
ESR 22mm/h,C反应蛋白(CRP)2.8 mg/L。
抗干燥综合征抗原A(SSA)抗体80.77 RU/ml,其他抗体检测结果均为阴性。血清1,3-β-D葡聚糖(G)试验、血清半乳甘露聚糖(GM)试验、T-SPOT、痰抗酸杆菌涂片、痰培养、支气管肺泡灌洗液二代测序检测结果均为阴性。支气管镜检查未见明显异常,肝、肾功能正常。
肺功能检查见表1;影像学检查见图1。
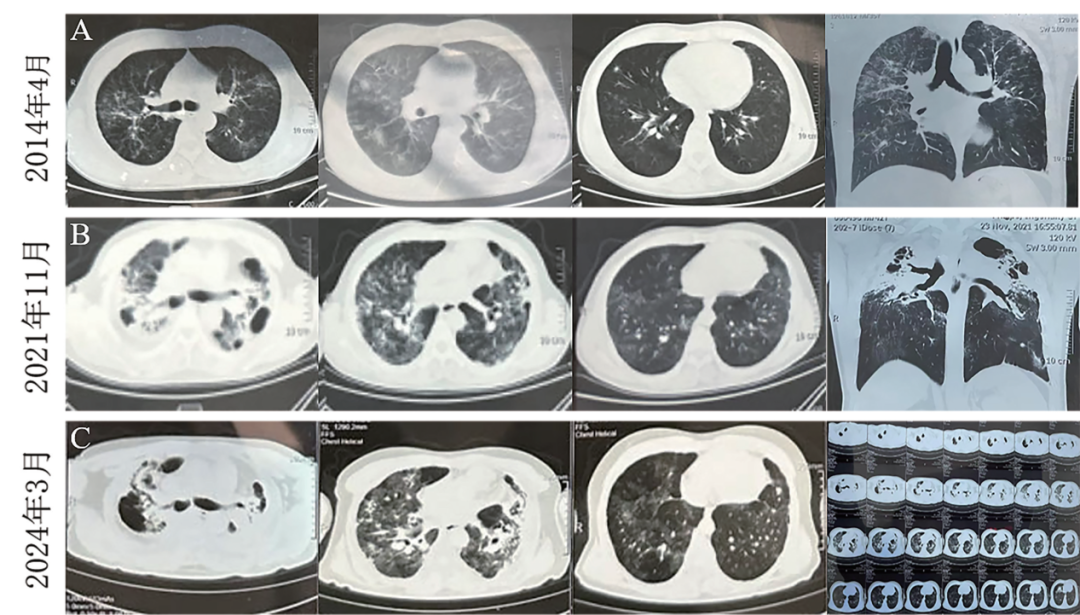
图1 患者不同时期胸部CT变化情况
A.2014年4月双肺可见散在斑片状密度增高影及小腺泡影,双上肺为著;B.2021年11月,两肺间质纤维化伴感染,双肺肺气肿,上叶肺大疱,可能伴有支气管扩张;C.2024年3月,两肺间质性改变并感染形成,双肺上叶部分实变,双侧肺门上提,双侧胸膜增厚、粘连
患者临床诊断为特发性胸膜肺实质弹力纤维增生症(IPPFE)。诊断明确后给予吡非尼酮治疗,我们将继续随访、跟踪,以明确患者对于上述治疗方案的反应。
本例患者为44岁男性,从最初发现胸部影像学异常至今10年余,患者的临床特点包括:①无其他系统慢性疾病史,无药物、毒物摄入史;②临床表现主要为进行性加重的干咳、活动后气短,查血常规、T-SPOT、肿瘤系列、自身抗体、痰液相关检查均阴性,抗感染、对症治疗无效;③最近一次的胸部CT提示双肺间质性改变,双肺上叶体积缩小,双肺上叶部分实变,其内空洞,支气管牵拉扩张,双侧肺门上提,双侧胸膜增厚、粘连;④肺功能表现为重度混合性通气功能障碍,中度弥散功能障碍,RV升高,RV/TLC %pred≥115%;⑤可排除结核性胸膜炎、过敏性肺炎及继发性PPFE等疾病。总结患者的临床特点,符合无外科肺活检(SLB)的IPPFE临床诊断标准。本例患者存在长期吸烟史,不能排除合并慢性阻塞性肺疾病,但单纯慢性阻塞性肺疾病无法解释上述病情特点。值得一提的是,研究表明环境暴露可能是IPPFE的诱因之一。
该患者外院诊断为陈旧性肺结核,接受了为期一年的抗结核治疗,但症状和胸部CT均无好转。IPPFE的临床症状无特异性,影像学可见肺顶端局灶性纤维化,这些表现容易被认为是结核感染的结果,尤其是在肺结核发病率较高的地区。结核病患者通常有结核中毒症状,结核性胸膜炎胸腔积液吸收可形成广泛胸膜增厚,胸膜组织见干酪样坏死、抗酸染色阳性或培养结核分枝杆菌阳性可确诊,抗结核治疗有效。本例患者无结核病史,无结核中毒症状,胸膜增厚以对称性上胸部分布为主,不符合结核性胸膜炎胸膜增厚的特点,并且本例结核相关检查均无阳性提示,所以可以排除陈旧结核感染的可能。除肺结核外,还要注意IPPFE与非特异性间质性肺炎(NSIP)、结缔组织疾病相关性肺病、结节病及石棉肺等疾病的鉴别。NSIP一般为下肺受累,肺组织可呈蜂窝样改变,胸膜增厚不明显;结缔组织疾病中,强直性脊柱炎肺受累时可表现为上肺纤维化,但一般不伴有明显的上胸部胸膜病变;结节病晚期可出现上肺纤维化、胸膜增厚,但结节病组织学主要表现为肉芽肿性病变,且具有较明确的病史;石棉肺一般有石棉、粉尘接触史,胸膜病变常伴有钙化,病灶没有明显的上胸部分布为主的特征。
目前,临床上IPPFE的治疗以对症支持治疗为主,药物治疗通常选用小剂量糖皮质激素,部分合用免疫抑制剂,也有应用抗纤维化药物吡非尼酮、尼达尼布的报道,但多项研究提示激素及免疫抑制剂对该疾病治疗效果差,关于抗纤维化药物的治疗效果也是说法不一,疾病进展至终末期可选择肺移植。本例患者诊断明确后给予吡非尼酮治疗,我们也将继续随访患者对于上述治疗方案的反应。
IPPFE是间质性肺疾病的一种罕见亚型。近年来,随着临床上对IPPFE的认识逐渐加深,其患病率显著增加,有研究显示,在特发性间质性肺炎(IIP)中有7.7%(29/375)的患者确诊了IPPFE。然而,关于IPPFE的病因和发病机制尚不清楚,可能与环境暴露、免疫功能失调和遗传易感性等因素密切相关。截至目前的循证医学证据显示,IPPFE可于13~87岁发病,就诊年龄呈双峰分布,高峰较早出现在30岁左右,较晚出现在60岁左右,无明显性别差异。较常见的临床表现包括随时间进展而逐渐加重的干咳、劳力性呼吸困难、胸部不适和体重减轻,其中30%的患者可发生单侧或双侧自发性气胸。IPPFE患者通常体型较消瘦,查体杵状指少见,部分患者可有扁平胸廓(胸廓前后径与胸廓横径比值减小),胸骨上切迹加深。听诊的呼吸音可能正常,当肺部病变扩展到上叶以外的部分或合并其他间质性肺疾病时,听诊两肺呼吸音减弱,或可闻及吸气相啰音。IPPFE患者的肺功能检查通常表现为限制性通气功能障碍,FVC显著下降,TLC下降,RV相对增加,RV/TLC比值增加。RV/TLC比值增加在其他IIP中并不常见,是一个可用于IPPFE诊断的显著特征。FVC的下降有两种不同的模式:短期快速下降和长期缓慢下降。在疾病的早期阶段,动脉血气分析通常显示PaO2、肺泡-动脉氧分压差(PA-aDO2)正常,PaCO2轻度升高,6分钟步行试验(6MWT)很少出现SaO2降低的情况。这些血气特征不同于以PaO2、PaCO2降低,PA-aDO2升高和6MWT过程中SaO2降低为血气特征的其他间质性肺疾病,对IPPFE的诊断和评估可有一定提示意义。随着疾病进展,晚期可出现低氧血症伴高碳酸血症性呼吸衰竭,慢性呼吸衰竭是这类患者死亡的主要原因。部分病例中多种血清自身抗体轻度升高,这可能与机体免疫功能失调有关。
IPPFE的典型组织病理学特征是双肺上叶脏层胸膜显著纤维化导致胸膜增厚,邻近胸膜下区域肺泡内纤维化、肺泡间隔弹性纤维增生。病变部位与相邻正常肺组织分界明显,离胸膜较远的肺实质不受累或受累轻微。IPPFE患者肺下叶常合并其他类型的纤维化,其中35%~50%为合并普通型间质性肺炎,随着IPPFE的进展,下叶纤维化也会逐渐加重。15%~35%的患者存在肉芽肿,一般认为是对感染性或非感染性抗原的生物反应引起,与IPPFE患者更好的预后相关。
IPPFE具有独特的胸部影像学特征。胸片典型表现为双上肺胸膜增厚,肺门上提。胸部高分辨率(HR)CT在IPPFE的诊断中具有重要作用,疾病早期一般表现为双侧肺尖胸膜下网格、小结节影;进展期主要表现为双上肺胸膜不规则弥漫增厚,胸膜下肺组织纤维化病变呈条索影,双侧肺门牵拉性上移;晚期IPPFE可见支气管牵拉扩张、肺组织囊性变、肺大疱。
目前,IPPFE的诊断尚无统一标准,应根据临床、影像、病理结果综合得出。一方面,肺组织病理学检查是IPPFE确诊的最终标准,但由于SLB是一项有创性的操作,可导致出血、气胸等严重并发症,临床上很少有机会获得患者的组织病理学资料。另一方面,IPPFE尚缺乏有效治疗手段,且30%的患者易并发难治性气胸,因此对这部分患者进行SLB可能弊大于利。在许多病例中,IPPFE的诊断与评估着重基于HRCT的结果。
2019年,瓦塔纳贝(Watanabe)等人提出无SLB的IPPFE诊断标准。
(1)可能符合放射学的IPPFE 表现为双上肺多部位胸膜下纤维化伴牵拉性支气管扩张。
(2)符合放射学的IPPFE 包括①出现干咳、劳累性呼吸困难等呼吸道症状,起病隐匿;②胸部X线显示双侧肺门结构上提和(或)HRCT显示双上肺多部位胸膜下纤维化伴牵拉性支气管扩张、上叶肺体积缩小;③对于IPPFE,应排除放射学和(或)组织学特征符合PPFE的其他病因,如慢性过敏性肺炎,以及结缔组织疾病、肺移植或造血干细胞移植等相关病因引起的继发性PPFE。
(3)符合放射学和生理学的IPPFE 生理学的标准包括RV/TLC %pre≥115%或 BMI≤20 kg/m2且RV/TLC %pred≥80%。若病变累及下肺,应经多学科会诊讨论确诊。本例患者即为临床诊断的IPPFE。苏祖基(Suzuki)等人的研究表明,临床诊断的IPPFE与病理诊断的IPPFE的临床特征无差异。
IPPFE患者预后较差,据报道5年生存率仅为23.3%~58.9%,影响预后的因素包括高龄、男性、合并下叶间质性肺炎和低FVC。然而,目前尚无明确有效的药物治疗方法,多数患者确诊后经验性使用免疫抑制剂、类固醇或预防性抗生素进行治疗,但临床症状均无缓解,肺功能获益甚微。有研究表明,吡非尼酮可延缓这类患者肺功能的下降,可能具有潜在的治疗意义。已有几例成功肺移植的病例报道,可作为疾病进展至终末期的治疗选择。关于IPPFE的治疗仍需要进一步的临床研究。
IPPFE具有独特的临床、影像学和病理学特征,有时与其他间质性肺疾病合并存在,临床中对于以肺上叶纤维化为主的患者,应注意与其进行鉴别,以减少漏诊和误诊。随着认识的不断加深,IPPFE病例的发现越来越多,这表明它可能并不像以前认为的那样罕见,但目前尚无有效的治疗方法。临床医生应进一步加强对该疾病的认识,帮助更多的患者得到及时的诊断和治疗,这也有助于我们开展进一步的队列研究来更好地探究该疾病的不同表型和评估现有治疗模式的效果,从而提高诊断和治疗IPPFE的能力。


本文由中国医学论坛报社呼吸与危重症编委会编委
空军军医大学西京医院宋立强教授组稿
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017