


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




概述
新生儿糖尿病(neonatal diabetes mellitus,NDM)是指出生后6 个月内出现的一种罕见的单基因糖尿病,也有部分 NDM 在出生 6 个月后发病的病例报道。常以糖尿病酮症酸中毒或血糖明显升高起病。该病可进一步细分为永久性新生儿糖尿病(permanent neonatal diabetes mellitus,PNDM)和暂时性新生儿糖尿病(transient neonatal diabetes mellitus,TNDM)。
病因和流行病学
NDM 呈常染色体显性、隐性或非孟德尔遗传。其病因为胰岛β细胞发育、功能或胰岛素信号通路中起关键作用的单个基因突变造成胰岛β细胞缺失或功能丧失而致病。目前已发现 23 种不同的 NDM 临床亚型,包括染色体6q24 区印迹异常及 22 个基因突变:KCNJ11、ABCC8、INS、GCK、ZFP57、SLC19A2、GATA6、GATA4、SLC2A2、HNF1β、PDX1、PTF1A、EIF2AK3、MNX1、NEUROD1、NKX2-2、IER3IP1、FOXP3、GLIS3、NEUROG3、RFX6、STAT3,每种亚型均有其特征性临床表现和遗传方式。其具体突变基因、发病机制、临床表现及推荐治疗方法见表 80-1。其中,6q24 区印迹异常是造成 TNDM 最常见的致病原因。6q24 区包含两种基因:PLAGL1 和 HYMAIM,其通过以下 3 种方式过表达而致病:①父源单亲二体型;②父源 6 号染色体不平衡重复;③母源 6q24 区低甲基化。该印迹基因是垂体腺苷酸环化酶激活多肽(胰岛素分泌的重要调控因子)1 型受体的转录调节因子,有研究显示其突变是 TNDM 的主要原因。编码ATP 敏感性钾离子通道(KATP)Kir6.2 亚单位的 KCNJ11 基因和磺脲类受体 1 亚单位(SUR1)的ABCC8基因及 INS 基因是导致 PNDM 最常见的致病原因。KCNJ11 基因和ABCC8 基因激活突变时,KATP通道对细胞内 ATP/ADP 比例变化不敏感,在葡萄糖刺激下通道无法正常关闭,细胞膜持续处于超极化状态,细胞外Ca 2+无法内流,造成胰岛素无法正常释放导致高血糖;INS 基因突变可导致胰岛素原分子的错误折叠并聚集于内质网,引起内质网应激和β细胞凋亡导致高血糖。NDM 的发生率为 1/500 000~1/400 000,但也有研究报道意大利、德国等发病率稍高,我国尚无相关数据报道。
临床表现
TNDM 常表现为严重的宫内发育迟缓,出生后很早(常在出生后1 周)即出现严重的、非酮症性高血糖,12 周后可恢复,50%~60%在青春期前后复发,复发后临床表现类似于 2 型糖尿病(type 2 diabetes mellitus,T2DM)。PNDM 多为出生时小于胎龄儿,常以糖尿病为唯一临床表现,也有少部分患者同时具有胰腺外的临床特征,如 NEUROD1 基因突变致PNDM常伴有中枢神经系统异常;HNF1β基因突变致 PNDM 常伴有肾脏病变或生殖系统异常等。PNDM无缓解期。
辅助检查
1.实验室检查:血糖、胰岛素、C 肽水平;尿常规(尿酮体、尿糖),血渗透压,血电解质;NDM 患者常以糖尿病酮症酸中毒起病,有血糖水平明显升高,尿酮体、尿糖阳性;1 型糖尿病抗体,NDM 患者 1 型糖尿病抗体阴性。
2.遗传学检测:NDM 已知致病基因的 Panel 检测;全外显子测序。
诊断
新生儿糖尿病的诊断依靠临床表现、实验室检查和基因检测。出生后6 个月内出现高血糖相关临床表现,结合实验室检查提示血糖升高的患者需进行基因检测。基因检测可明确大约 80%的 NDM 患者。具体分型和确诊需依靠基因检测确定。高通量测序可同时完成多个基因检测,极大提高了临床表型类似的NDM的诊断效率。
鉴别诊断
NDM 需与 1 型糖尿病、早发 2 型糖尿病及其他单基因糖尿病相鉴别,如青少年发病的成人型糖尿病(maturity onset diabetes of the young,MODY)等,1型糖尿病抗体检查及基因检测对于病因的鉴别具有重要意义。主要的考虑因素如下:
1.年龄:多数 PNDM 为出生后 6 个月内发病,TNDM 为出生后1 周内发病;1 型糖尿病及 MODY 发病年龄相对较晚。
2.家族史:MODY 患者多有三代或三代以上家族遗传史,可与NDM相鉴别。
3.实验室检查:1 型糖尿病抗体检测,NDM 患者抗体为阴性;基因检测可用于区分 NDM 的具体亚型,并与 MODY 进行鉴别。
治疗
不同 NDM 亚型治疗原则不同,需具体亚型具体分析。TNDM患儿发病后可使用胰岛素治疗,且用量可迅速减少,经过平均 12 周后即可不再需要治疗;但该病多于青春期前后复发(复发率为 50%~60%),复发后临床表现类似于早发T2DM,表现为第一时相胰岛素分泌缺失,对磺脲类药物有反应,不一定需要胰岛素治疗。约 90%的 KATP通道基因(KCNJ11 和 ABCC8)突变致PNDM患者可使用磺脲类药物治疗。磺脲类药物可改善该类患者的血糖控制,且不增加低血糖风险。与成年 T2DM 患者相比,这类 PNDM 患儿常需较高剂量的磺脲类药物,以目前临床应用最多的格列本脲为例,平均用量为 0.5mg/(kg·d),最大剂量可达 2.3mg/(kg·d)。其余类型 PNDM,胰岛素仍是唯一选择,与1 型糖尿病不同的是,这类 PNDM 患者胰岛素起始剂量偏大,为 0.5~1.2U/(kg·d)。
诊疗流程(图80-1)
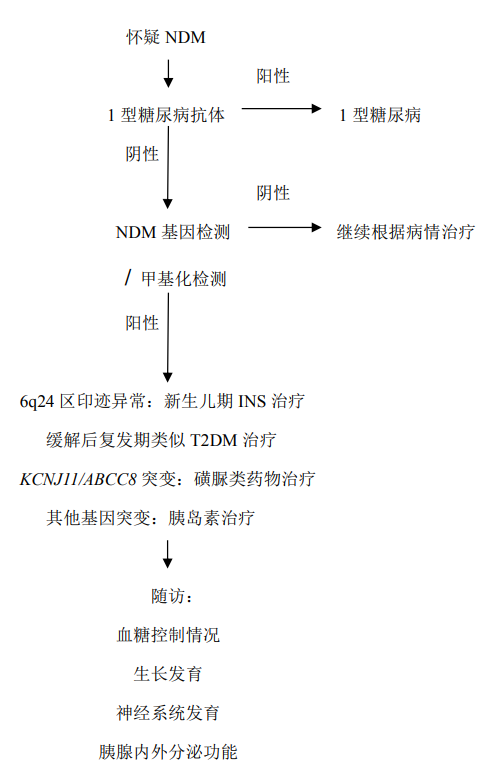
图 80-1 新生儿糖尿病(NDM)诊疗流程
表80-1 NDM亚型

参考文献(略)
来源:国家卫生健康委员会《罕见病诊疗指南(2019年版)》
罕见病诊疗指南——黏多糖贮积症【心肾系统罕见病】
罕见病诊疗指南——甲基丙二酸血症【神经系统罕见病】
罕见病诊疗指南——羟化酶缺乏症【心肾系统罕见病】
罕见病诊疗指南——Gitelman综合征
罕见病诊疗指南——抗 LGI1 抗体相关脑炎【神经系统罕见病】
罕见病诊疗指南——遗传性痉挛性截瘫【神经系统罕见病】
罕见病诊疗指南——视神经脊髓炎【神经系统罕见病】
罕见病诊疗指南——淋巴管肌瘤病【罕见肿瘤】
罕见病诊疗指南——自身免疫性胰岛素受体病【免疫系统罕见病】
罕见病诊疗指南——原发性肉碱缺乏症【儿童罕见病】
罕见病诊疗指南——原发性肉碱缺乏症【儿童罕见病】
罕见病诊疗指南——β-酮硫解酶缺乏症【儿童罕见病】
罕见病诊疗指南——生物素酶缺乏症【神经系统罕见病】
罕见病诊疗指南——自身免疫性垂体炎【神经系统罕见病】
罕见病诊疗指南——心脏离子通道病【心肾系统罕见病】
罕见病诊疗指南——天使综合征【神经系统罕见病】
罕见病诊疗指南——自身免疫性脑炎【神经系统罕见病】
罕见病诊疗指南——抗NMDAR脑炎【神经系统罕见病】
罕见病诊疗指南——白化病【呼吸和皮肤系统罕见病】
罕见病诊疗指南——精氨酸酶缺乏症【神经系统罕见病】
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017