


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:东部战区总医院 国家肾脏疾病临床医学研究中心 焦晨峰 范文静 梁少姗 张炯
病史摘要
主诉 患者为12岁女性,因“发现尿检异常6年”于2023年8月21日入院。
现病史 2017年8月,患者海边游泳后出现发热及皮疹(背部、四肢出现充血性斑丘疹),于当地医院就诊,查尿蛋白、隐血阳性,诊断为“猩红热、化脓性扁桃体炎”,给予抗生素静滴治疗,发热及皮疹好转,但复查尿蛋白持续阳性。2017年10月,患者至某儿童医院就诊,查尿蛋白3+,尿红细胞8~12 /HP(非均一型),肾功能正常,未行肾活检,予培哚普利、阿魏酸哌嗪、维生素AD等药物治疗,后定期复查24 h尿蛋白定量波动在1 g左右。2020年11月,患者再次就诊于某儿童医院行有机酸代谢缺陷筛查,血同型半胱氨酸升高,尿甲基丙二酸和甲基枸橼酸升高。诊断“甲基丙二酸血症伴高同型半胱氨酸血症”,予羟钴胺肌注,左卡尼汀、甜菜碱、叶酸、福辛普利口服治疗2年,后羟钴胺肌注切换为甲钴胺口服,停用左卡尼汀和甜菜碱;2023年3月停用所有药物。停药期间复查尿蛋白2+~3+。患者目前精神可,体力及食欲正常,排尿正常,大便正常,近2年体重增加约15 kg。为行进一步诊治入院。
既往史 患者1岁半学会走路,大动作和精细动作及计算能力较同龄人差,一般沟通交流正常,语言表达、记忆力较好。自幼无发热惊厥、癫痫、食欲减退、生长缓慢、运动倒退、痉挛步态等表现;半年前发现血压偏高,最高140~150/80~90 mmHg;双眼近视4年余,约400 °;有“过敏性鼻炎”病史;余无特殊。
月经史 12岁初潮,5 d/30 d,末次月经时间2023年8月10日,经量正常,颜色正常,无痛经,经期规律。
家族史 父母非近亲结婚,母亲妊娠期间无特殊药物使用史及患病史。否认家族遗传病及类似疾病史。
体格查体
体温36 ℃,脉搏80 次/min,呼吸18 次/min,血压117/89 mmHg,身高164 cm,体重72.6 kg,体质指数27 kg/m2,发育正常,心肺腹部查体未及异常,双下肢无水肿。
实验室检查
尿液 尿蛋白定量 1.51 g/24 h;红细胞计数7.2 /HP(非均一型),尿C3 2.07 mg/L,尿α2-巨球蛋白(α2-M) 2.00 mg/L;尿N-乙酰-β-D-氨基葡萄糖甘酶(NAG)21.5 U/(g·cr),视黄醇结合蛋白(RB蛋白)0.50 mg/L;尿甲基丙二酸 70.8(参考值0~4)。
血常规 白细胞计数10.79×109/L,血红蛋白138 g/L,平均红细胞容积100 fl,网织红细胞 2.88%,血小板计数338×109/L,红细胞碎片阴性,C反应蛋白<0.5 mg/L。
生化 总蛋白58.9 g/L,白蛋白38.4 g/L,球蛋白20.5 g/L,尿素氮6.1 mmol/L,肌酐0.82 μmol/L,估算肾小球滤过率(eGFR)109 ml/(min·1.73m2),尿酸523 μmol/L,心肌损伤指标、肝功能及电解质、凝血功能、糖化血红蛋白、甲状腺功能、胰岛素样生长因子1、生长激素水平、血管内皮生长因子(VEGF)正常;甲状旁腺激素(PTH)23.45 pg/ml,总25羟维生素D 14.39 ng/ml;同型半胱氨酸137.5 μmol/L(参考值0~15 μmol/L),血氨79 μmol/L(参考值9~30 μmol/L);血清维生素B12 1064.0 pmol/L,血清叶酸 >23.50 ng/ml。
免疫学指标 抗核抗体(ANA)1∶256 , 抗双链DNA(ds-DNA)抗体、抗核抗体谱、抗磷脂酶A2受体抗体、免疫固定电泳、血游离轻链、补体、免疫球蛋白、淋巴细胞亚群、血管性血友病因子裂解酶(ADAMTS13)活性及抑制物未见异常。
辅助检查
双肾超声 左肾111 mm×50 mm×55 mm,右肾108 mm×43 mm×54 mm,皮质回声增强;双肾轮廓规则,包膜连续完整,双肾内未见肾盂肾盏扩张。
心脏超声 室间隔厚度7 mm,左心室舒张末内径36 mm,左室后壁厚度7 mm,超声心动图大致正常(左室射血分数62%),肺动脉收缩压13 mmHg。腹部及甲状腺超声未见异常。
其他 头颅核磁平扫未见异常;鼻中隔偏曲,左侧下鼻甲肥厚。眼底:正常,视乳头边界清,黄斑区正常,动静脉比例为2∶3。
肾活检病理
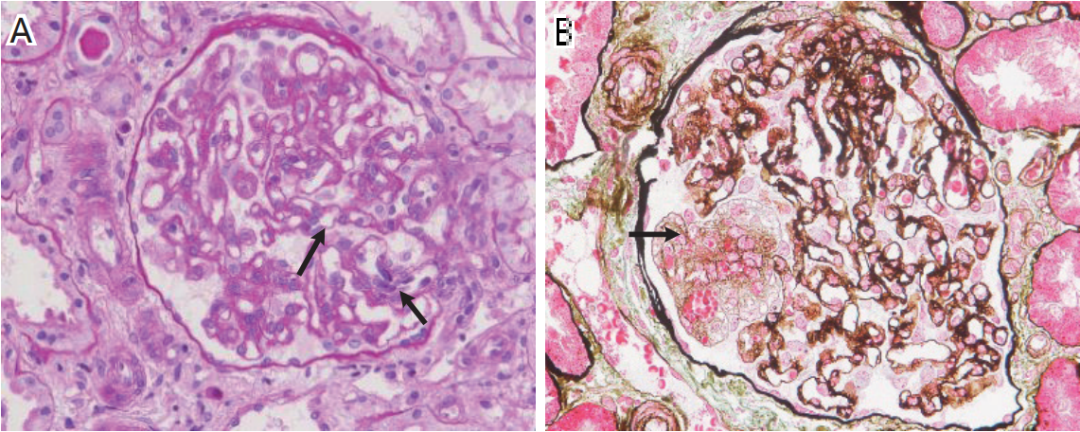
光镜下观察9个肾小球,2个球性废弃。余肾小球系膜区轻度增宽,系膜细胞和基质略增多,毛细血管袢开放好,节段袢腔内皮细胞增多(图1A),囊壁节段增厚分层。PASM-Masson染色见系膜溶解和外周袢融合呈瘤样扩张(图 1B),肾小球系膜区节段嗜复红物沉积,较多外周袢分层。肾小管间质轻度慢性病变(5%),灶性肾小管萎缩、基膜增厚,管腔内见蛋白管型,合并轻度急性病变(10%),灶性肾小管上皮细胞刷状缘脱落,灶性肾小管上皮细胞肥大伴管腔扩张,小管上皮细胞细颗粒变性,间质少量单个核细胞、偶见浆细胞浸润,灶性纤维化。动脉未见明确病变。
图1 肾活检病理
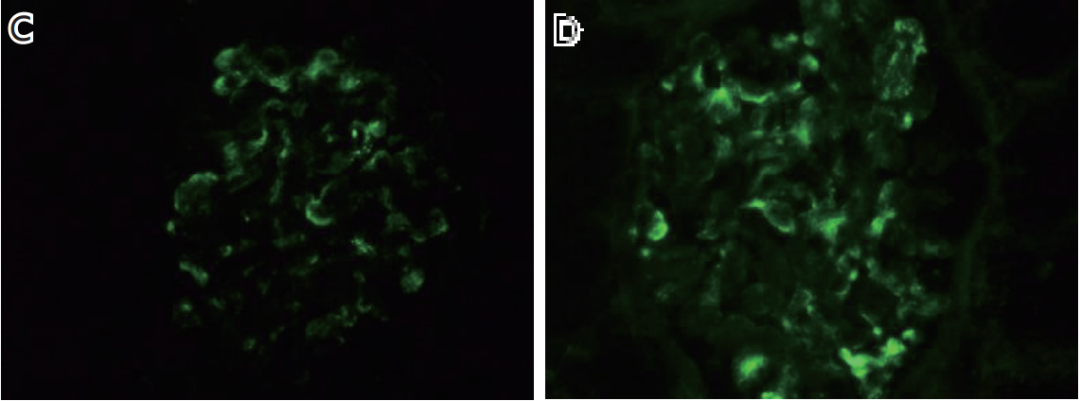
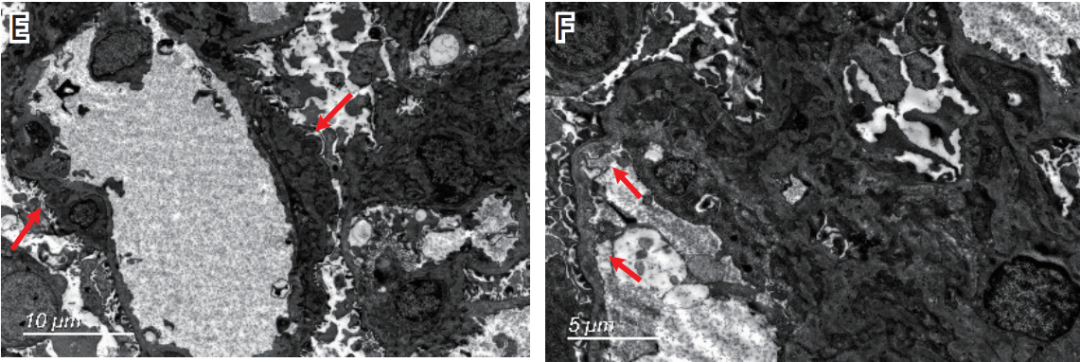
图1A 肾小球节段袢腔内皮细胞增多(↑),较多外周袢分层;图1B 肾小球节段系膜溶解和外周袢融合呈瘤样扩张(↑);图1C、图1D IgG++(C)、IgA+~++(D)弥漫颗粒状沉积于系膜区及血管袢;图1E 肾小球系膜区少量高电子密度的致密物沉积(↑),较多袢见细胞成分插入至基膜内皮下、新的基膜形成;图1F 肾小球可见基膜内皮下疏松(↑)。
免疫荧光免疫球蛋白(Ig)G++(图1C)、IgA+~++(图1D)、IgM ±、C3 ±、C1q ±、κ轻链+、λ轻链++,弥漫颗粒状沉积于系膜区及血管袢。IgG1+弥漫分布、IgG3 ±~+节段分布,颗粒状沉积于系膜区及血管袢。IgG2、IgG4阴性。
电镜下观察2个肾小球。肾小球系膜区增宽,系膜细胞和基质增多,系膜区少量高电子密度的致密物沉积(图1E),偶见系膜区结构疏松。肾小球毛细血管袢开放好,较多袢见细胞成分插入至基膜内皮下、新的基膜形成,数处基膜内皮下疏松(图1F)、区域增宽,基膜内皮下散在电子致密物沉积,上皮侧未见电子致密物沉积。肾小球足细胞足突节段融合,约10%~20%。
病理诊断:① 血栓性微血管病(TMA);② 免疫复合物介导的肾小球肾炎。
基因检测
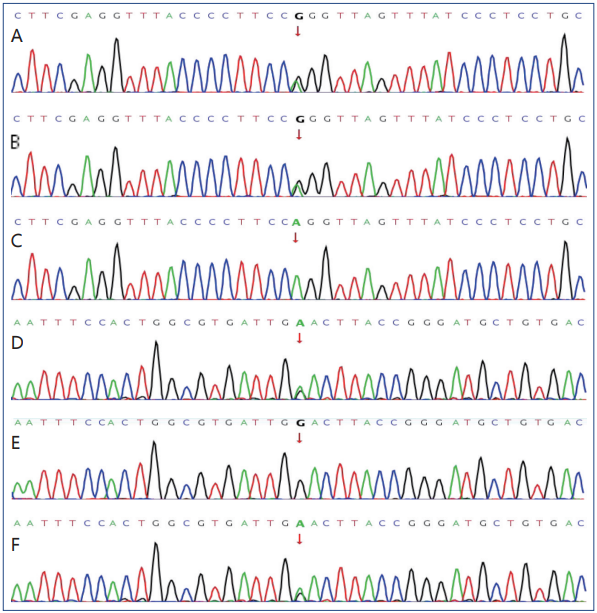
经患者及家属同意,抽取患者外周血行全外显子基因测序,并抽取父母外周血做验证。证实患者存在MMACHC基因c.80A>G(p.Gln27Arg)和c.609G>A(p.Trp203Ter) 2个杂合子变异,该变异为复合杂合突变。c.80A>G(p.Gln27Arg)位点母亲未见突变,父亲为杂合突变。c.609G>A(p.Trp203Ter)位点母亲为杂合突变,父亲未见突变,说明先证者的这2个位点分别来自于父母(图2)。根据美国医学遗传学和基因组学学会(ACMG)的标准这2个变异为可能致病和致病性变异。
图2 患者及父母MMACHC基因检测结果
图2A 患者MMACHC基因 c.80A>G突变;图2B 患者父亲MMACHC基因 c.80A>G突变;图2C 患者母亲MMACHC基因 c.80A>G未见突变;图2D 患者MMACHC基因c.609G>A突变;图2E 患者父亲MMACHC基因c.609G>A未见突变;图2F 患者母亲MMACHC基因c.609G>A突变
诊断与治疗
患者诊断为:① 甲基丙二酸血症(MMA)MMACHC基因c.609G>A及c.80A>G复合杂合突变);② TMA;③ 免疫复合物介导肾小球肾炎。
患者出院后给予甲钴胺(5 mg,3次/d)、叶酸(10 mg/d)、左卡尼汀、甜菜碱(3 g,3次/d)、福辛普利等治疗。2024年1月,当地医院复查尿蛋白695.4 mg/24h,尿红细胞阴性,SCr70.8 μmol/L,同型半胱氨酸 126 μmol/L,血氨正常。
诊疗分析
患者为青少年女性,病程6年,临床表现为慢性肾炎综合征,中等量蛋白尿,伴镜下血尿,血压高,无贫血,肾功能正常,肾活检病理提示TMA。肾损伤原因鉴别诊断如下:① 血清ANA低滴度阳性,抗ds-DNA抗体、抗核抗体谱、抗中性粒细胞胞质抗体(ANCA)均阴性,补体正常;肾外无光过敏、皮疹、关节痛、雷诺现象等表现,不支持自身免疫性疾病;② 血清ADAMTS13活性正常、抗体阴性,不支持血栓性血小板减少性紫癜;③ 血清补体水平正常,肾组织C3 trace沉积,基因检测未见补体旁路通道相关基因突变,不支持补体旁路异常活化所致肾损伤;④ 患者传染病四项阴性,近期无肺炎链球菌、大肠埃希菌等感染相关依据,实验室检查感染指标正常,不支持感染相关肾损伤;⑤ 患者非肿瘤好发年龄,查血游离轻链正常、免疫固定电泳阴性,全身未见多发淋巴结、肝脾肿大,不支持肿瘤相关肾损伤。结合患者既往有甲基丙二酸血症,基因检测提示MMACHC基因复合杂合突变,考虑MMA相关TMA。
讨论
MMA的临床表现及分类
MMA是我国最常见的有机酸血症,呈常染色体隐性遗传,由于甲基丙二酰辅酶A变位酶(MCM)自身缺陷或其辅酶钴胺素(cbl)代谢障碍,造成患儿体内甲基丙二酸、丙酸、甲基枸橼酸等代谢物异常蓄积而导致的代谢紊乱及多器官损伤,包括神经、肝脏、肾脏、血液、心肺等。临床表现为喂养困难、生长发育延迟、小头畸形、癫痫、慢性肾脏病、巨幼细胞性贫血、肥厚型心肌病、肺动脉高压等。根据是否伴有高同型半胱氨酸血症,可分为单纯型MMA及伴高同型半胱氨酸血症的合并型MMA。后者约占我国患者的70%。本例患者出生后出现喂养困难及神经系统发育障碍,后续出现肾脏损伤,尿有机酸筛查提示尿甲基丙二酸水平升高,血同型半胱氨酸水平升高,明确诊断为合并型MMA。
人体内钴胺素的合成
自然界的cbl由微生物合成,高等动植物无法自身合成。人体需要摄入动物性食物补充cbl。进入细胞质中的cbl要经cblC(MMACHC基因编码)和cblD(MMADHC基因编码)加工、修饰和分化,形成甲钴胺和羟钴胺。甲钴胺在细胞质中作为甲硫氨酸合成酶的辅酶,催化同型半胱氨酸转化为甲硫氨酸。羟钴胺进入线粒体,作为MCM的辅酶,催化甲基丙二酰辅酶A转化为乙酰辅酶A,最终进入三羧酸循环。目前已知至少有21个基因参与人体cbl的吸收、存储和代谢。cblC是维生素B12加工蛋白,cblC缺陷,甲基丙二酰辅酶A不能进入三羧酸循环,导致甲基丙二酸体内蓄积及同型半胱氨酸水平升高。
MMA肾脏损伤表现
MMA导致的肾脏损伤主要表现为TMA,此外包括肾小管间质性肾炎、肾小管酸中毒、膜增生性肾小球肾炎、膜性肾病。本例患者肾穿刺活检提示TMA,排除其他继发因素包括感染、自身免疫性疾病、肿瘤、补体旁路活化、血栓性血小板减少性紫癜等,结合患者既往MMA病史,完善基因检测提示MMACHC基因突变,最终考虑MMA导致TMA。MMA导致TMA具体机制尚不清楚,可能与高同型半胱氨酸减少血管内皮一氧化氮的产生、增强氧化应激、刺激血管平滑肌细胞增生、改变血管壁弹性损伤血管内皮细胞有关。血甲基丙二酸水平高损伤线粒体功能促进慢性肾脏病进展。代谢性酸中毒促进近端肾小管氨的产生、内皮素生成增多、激活补体旁路及肾素血管紧张素系统,加重慢性肾脏损害。然而,不伴MMA的高同型半胱氨酸血症如甲基四氢叶酸还原酶(MTHFR)或胱硫醚β合成酶(CBS)缺陷尚未见导致肾TMA相关报道;高同型半胱氨酸血症伴MMA的cbl其他缺陷类型如cblD、cblF、cblJ和cblX,导致TMA报告更为罕见。推测有其他因素参与TMA的发生。本例患者肾活检的特殊之处在于合并免疫复合物介导肾小球肾炎,目前文献未见相关报道。从一元论考虑与MMA相关,具体的发病机制不明。
cbl代谢障碍C型MMA的治疗
推荐羟钴胺补充钴胺素并最大限度地提高缺乏酶的活性,肌肉注射优于口服。叶酸和甜菜碱促进同型半胱氨酸代谢转化为甲硫氨酸。MMA常合并继发性肉碱缺乏,左卡尼汀与有机酸结合,形成水溶性代谢产物从尿液排出体外,促进有机酸的排泄。需要注意的是,cbl代谢障碍C型MMA患者的维生素B12水平并不缺乏。维生素B12水平正常并不是cbl功能缺陷可靠的指标。疾病预后与疾病类型、诊断早晚及长期合理治疗有关。部分患者经药物积极治疗后,肾功能明显改善,甚至摆脱透析。血浆置换及补体C5抑制剂依库珠单抗并不能改善此类患者TMA预后。本例患者维生素B12及叶酸水平正常,给予维生素B12甲钴胺替代治疗,经积极治疗后,生化指标改善,其长期预后有待随访进一步观察。
小结
肾脏TMA损伤病因多样,cbl代谢障碍C型MMA是其一种罕见的病因。对于多器官损害伴有高同型半胱氨酸血症合并肾脏疾病的早发年龄患者,要注意进行有机酸的筛查,并进一步行肾活检病理检查及基因检测明确诊断。早期识别并积极治疗,可有效改善患者预后。
END
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017