


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




李大伯(化名)今年快70岁了,但他却有着一段不同寻常的经历——从30多岁开始,他就感到四肢逐渐变得无力,这一症状并没有随着时间恢复,甚至持续了30多年。
尽管多年来李大伯辗转各地求医问药,但始终没有一个确切的诊断结果。年轻时,他主要是双下肢大腿无力,下蹲起立不能完成,但后来逐渐出现了双上肢无力的症状。2019年,李大伯还因为没有力气导致身体后仰摔倒过一次,并因此住了院,但未查明病因,还是不了了之。

随着日子一天天过去,李大伯的行动越来越不便,目前已不能独立行走,需要坐轮椅,日常起居都需要家人照顾。然而,李大爷从未放弃过寻找病因和治疗的希望。2023年,他曾经在当地一家三甲医院就诊,医院考虑他是肌肉病,并安排完善了全外显子基因检测,但仍无法明确诊断。
直到2024的春天,一次偶然的机会,李大伯的孙子小明(化名)通过网络了解到浙大二院医学遗传科在神经系统疑难杂症方面颇有研究,小明立刻带着爷爷来到了这家医院。
经过仔细查体和肌电图检查后,医学遗传科余昊医生怀疑李大伯患有一种罕见的神经肌肉疾病——脊髓性肌肉萎缩症(Spinal Muscular Atrophy,以下简称SMA)。这种疾病被称为“两岁以下婴幼儿头号致死性遗传病”,在全世界范围内SMA的发病率为1/6000~1/1,0000。该病在成人中更加罕见,因此也更容易被误诊漏诊。而且,这个基因突变的形式有些特殊,临床常用的全外显子测序有时会将其遗漏。余昊医生安排了特殊的针对性基因检测,最终确诊李大伯确实是脊髓性肌萎缩症。确诊了疾病之后,经过针对性的药物治疗,李大伯的无力症状得到了改善。
脊髓性肌肉萎缩症是一种常染色体隐性遗传病,是第一批国家罕见病目录中疾病之一。
这是一种神经肌肉病,患者会逐渐进行性、对称性的肌肉无力和萎缩,像软趴趴的玩偶一样,而后逐渐丧失包含呼吸、吞咽的各种运动功能(但智力、认知功能、感官系统等不受影响),最终导致卧床或死亡,给家庭和社会带来巨大的痛苦。值得一提的是,像上文提到的李大伯这样病程持续20~40年的成人SMA患者群体,曾被误诊为各种肌肉病的患者也不在少数。
SMA是一种隐性遗传疾病,必须要父母各带一个致病的等位基因,同时传给子女才能发病。在普通人群中,SMA致病基因的携带率非常高,约1/50,相当于50个人里面就有1个人可能携带致病基因。该病没有国界,不分人种,且不受年龄、种族或性别的限制,携带者也没有任何症状,因此很多家庭都是“意料之外”发现自己的孩子患病。
SMA根据发病年龄和疾病严重程度主要分为I型、Ⅱ型、Ⅲ型、Ⅳ型这4种临床类型。
I型发病年龄最早,对运动功能的影响最大,通常在6个月内发病、2年内致死;Ⅳ型则最轻,可以在成人期(20岁以后)发病,且不影响寿命。
婴幼年起病的SMA并不难诊断,但是这些成人型患者却经常面临“兜兜转转”,被各大医院长期误诊。自浙大二院创建科室以来,已有接近10例类似成人型SMA被诊断和接受治疗。
目前,浙大二院医学遗传科正在开展SMA筛查项目,若有怀疑该疾病的患者,可以前往我院就诊评估,若高度怀疑该病,可以免费进行相关基因检测。
罕见病向来治疗药物少、价格昂贵。SMA在经历了漫长无药可及的窘境后,迎来了治疗的新时期。在科学家的努力下,基因治疗药物开始走向临床。
相信大家一定之前一定看过2021年医保局“灵魂砍价”的视频,国家医保局谈判代表张劲妮,经过前后8轮谈判,将首次报价每支5万多元的药,价格降至3万多元并纳入医保目录。而上市之初,这款药物每支价格高达70万一支,被称为“天价”药。这个药物就是针对SMA研发的精准靶向药物——诺西那生钠注射液。此外,另一种靶向的口服药物利司扑兰口服溶液也已在国内获批,被纳入国家医保目录。
目前,医学遗传科有近50名不同年龄段的SMA患者会定期来院进行治疗。在专业的医生和护理团队保驾护航下,这部分患者的病情发展得到了控制。
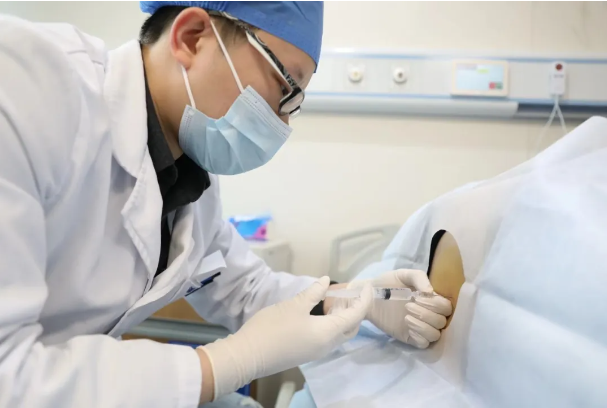
余昊医生正在为患者注射诺西那生钠注射液

浙大二院医学遗传科/罕见病诊治中心是国内首家医学遗传学临床科室,专门收治各种罕见病和遗传病。科室聚焦遗传病和疑难罕见病的规范化诊治,致力于从分子水平为疾病的早期诊断、遗传咨询、预防出生缺陷及综合治疗提供精准高效的新型医疗服务。病区设有独立的肌电图室、肌肉活检室,同时依托浙大二院医学遗传学实验室开展基因检测、肌肉病理、细胞和蛋白功能检测等技术,将极大提高疾病的诊断速度和诊断率,从而促进精准治疗。


本文转自:浙二吴志英
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017