



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




终末期肾病 (ESRD) 80% 的病因是蛋白尿性肾病。蛋白尿性肾病主要包括糖尿病肾病 (DKD)和局灶节段性肾小球硬化症 (FSGS),是一类以尿液中存在过量蛋白质为特征的疾病。其最终会进展为 ESRD,需要长期透析或肾移植治疗。脂质是维持足细胞生物学功能所必需的,而在蛋白尿性肾病中,足细胞会出现过多的脂质聚集,被称为足细胞脂毒性。
2024年3月,来自陆军军医大学第二附属医院的郑宏庭/郑怡团队在Advanced Science杂志发表题为 “Dock5 Deficiency Promotes Proteinuric Kidney Diseases via Modulating Podocyte Lipid Metabolism”的文章,揭示了蛋白尿性肾病中会出现胞质分裂因子 5 (Dock5)的缺失,并通过以 N6 甲基腺苷 (m6A)依赖的方式上调 LXRα 来增强 CD36 介导的足细胞脂肪酸摄取;恢复其表达可改善足细胞损伤和蛋白尿性肾病。研究结果表明Dock5 可能是足细胞脂毒性的关键调控分子,可作为蛋白尿性肾病的潜在治疗靶点。
作者首先筛选了可能参与足细胞脂质代谢的基因。利用钠-葡萄糖协同转运蛋白-2 抑制剂 (SGLT2i)改善足细胞脂毒性,分析SGLT2i 处理的足细胞的差异基因表达 (DGE) 数据集;利用 DKD 小鼠模型的足细胞分析蛋白尿性肾病的足细胞 DGE 数据集。综合分析这两个数据集的交集并确定了 15 个相关基因,其中的8 个基因在疾病模型和 SGLT2i 治疗间呈现出相反的变化趋势,而Dock5 在足细胞中的表达最高。
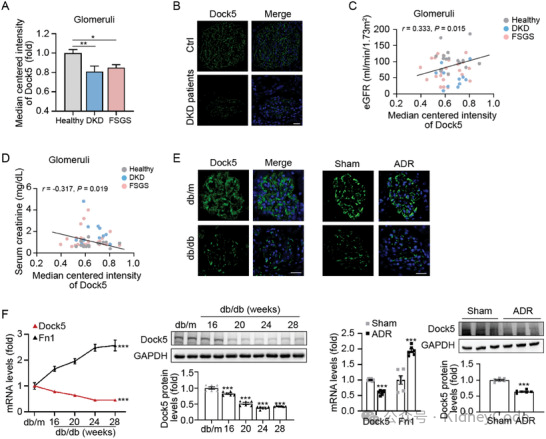
接下来,作者在DKD/FSGS患者和小鼠模型(db/db 诱导的 DKD 小鼠和 ADR 诱导的FSGS 小鼠)中观察 Dock5 的表达。发现患者的Dock5 表达显著降低,并与 eGFR 正相关,与Scr水平负相关(图1)。小鼠模型中Dock5 表达与患者的观察结果一致(图1)。以上结果表明 Dock5 可能参与蛋白尿性肾病的进展。
图1 Dock5 在蛋白尿性肾病患者和小鼠中表达降低
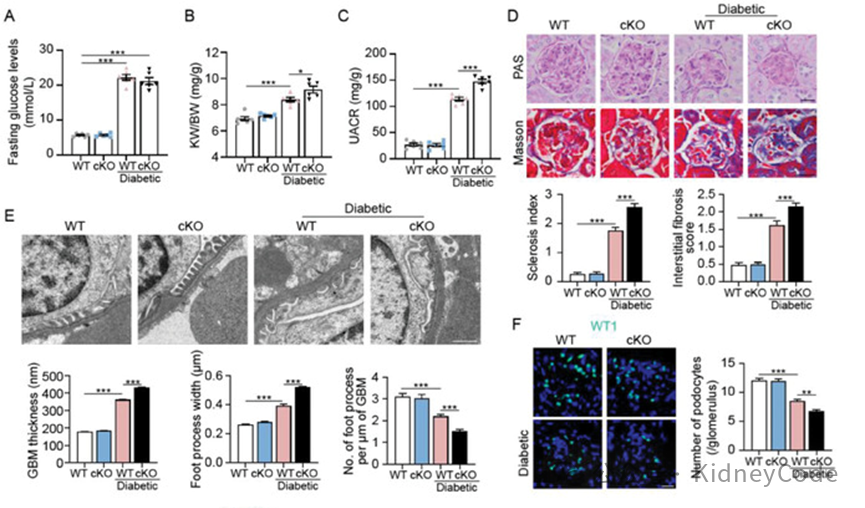
随后,作者构建足细胞特异性 Dock5 敲除小鼠(Dock5fl/fl‐Cre+,cKO),发现在诱导 DKD 发病后,cKO 小鼠的尿白蛋白与肌酐比(UACR)升高,肾小球病理程度加重,肾小球基底膜增厚,足细胞足突增宽并消失,足细胞丢失增多(图2)。FSGS小鼠也观察到类似结果。以上数据表明,Dock5 缺失会在蛋白尿性肾病进展过程中加剧足细胞和肾小球损伤。
图 2 足细胞中 Dock5 缺失加剧了蛋白尿性肾病的肾损伤
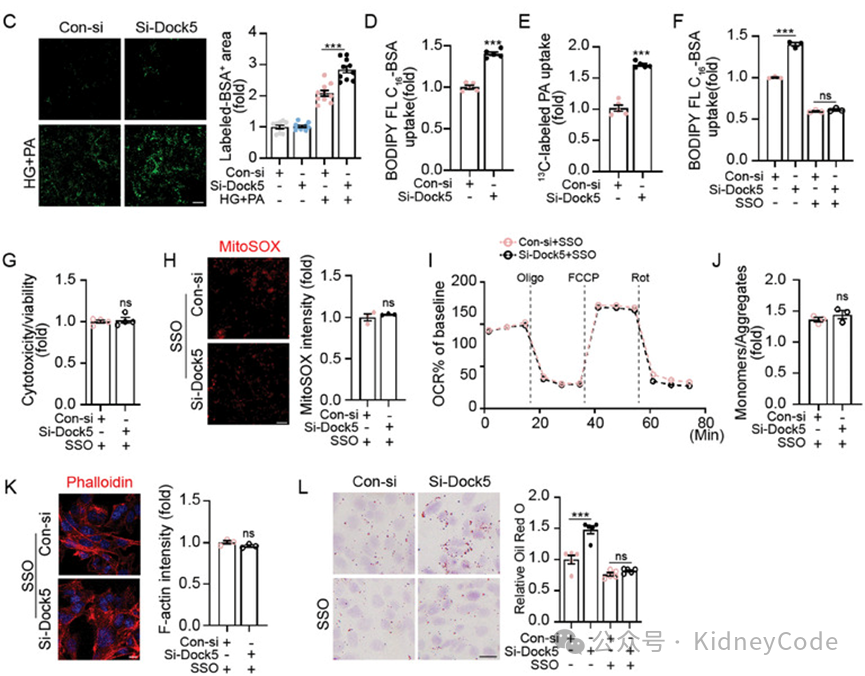
诱导 DKD 后的 cKO 小鼠中观察到足细胞出现严重的脂质积累。利用脂质组学分析,作者进一步发现脂质沉积的主要种类是游离脂肪酸等,而非胆固醇酯和磷脂酰胆碱,这表明Dock5缺失主要影响脂肪酸代谢。脂肪酸代谢包括摄取、合成和氧化过程,因此作者在 si‐Dock5 的足细胞中对这些过程进行了测量,发现Dock5的敲低显著提高了脂肪酸的摄取。摄取主要通过两种途径:一种是通过GPCR介导的内吞或巨胞饮作用;另一种是CD36依赖的蛋白介导的运输。
Dock5 敲低未改变脂质结合GPCR 的水平;而 CD36 抑制剂 SSO 消除了Dock5 缺失引起的脂肪酸摄取。这表明 Dock5 缺失主要通过 CD36 调节蛋白介导的途径增强脂肪酸的摄取,从而诱发足细胞损伤。(图3)
图3 Dock5 缺失促进 CD36 介导的脂肪酸摄取
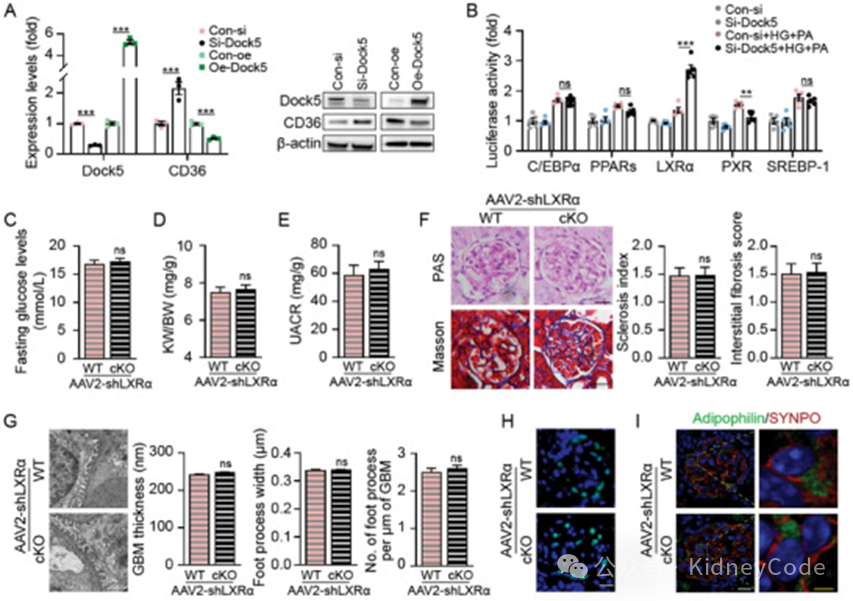
作者进一步研究了Dock5调控CD36的机制。由于CD36的mRNA水平受Dock5调控,提示这一调控过程可能发生在转录水平上。一些转录因子被认为是 CD36 转录的重要调节因子,作者分别过表达相关转录因子,发现只有 LXRα 表达后,Dock5 敲低会增加报告基因活性。在 DKD 发病条件下,通过 AAV2 ‐Nphs1‐shLXRα 对 Dock5 cKO 小鼠的足细胞进行 LXRα 敲低,此时 Dock5 缺失引起的病理改变(UACR、足细胞足突增宽和消失、足细胞丢失和脂质积聚等)显著减弱(图4)。LXRα 拮抗剂 GSK2033 消除了Dock5 敲低诱导的脂肪酸摄取、抑制了CD36 及其下游基因表达,减轻了足细胞系中的脂毒性。进一步探究发现LXRα 通过直接结合足细胞中的启动子区的DR-7 型核受体反应元件,激活了 CD36 基因转录。这些结果表明 Dock5 主要通过转录因子 LXRα 调节 CD36表达。
图 4 Dock5 通过 LXRα 调节 CD36 介导的脂肪酸摄取
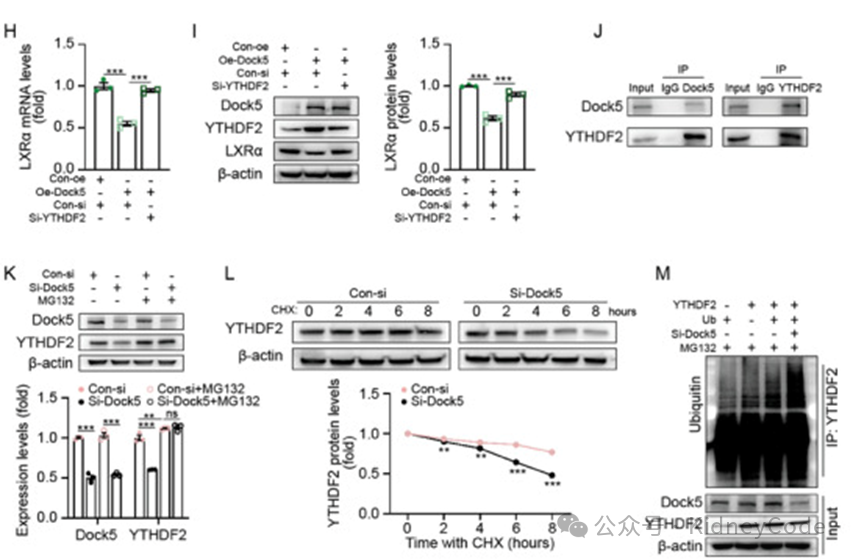
接下来,作者探究了 Dock5 调控 LXRα 的机制。由于Dock5 缺失没有改变LXRα 前 mRNA的丰度,因此作者猜测其水平可通过 m6A介导的 mRNA 衰减进行调节。LXRα的成熟 mRNA 可与 m6A 读取蛋白 YTHDF2 相互作用诱导 mRNA 衰变。RNA 免疫沉淀分析表明 YTHDF2与LXRα mRNA 结合。YTHDF2 敲低增加了 LXRα 蛋白和 mRNA 水平,而 m6 A 识别位点突变的 YTHDF2不能降低 LXRα 的水平。进一步在 Dock5 过表达的足细胞中敲低YTHDF2,发现原本被Dock5 抑制的 LXRα 表达增加(图5)。随后作者验证发现 DOCK5 与 YTHDF2 存在相互作用。Dock5 过表达后 YTHDF2蛋白水平升高 ,敲低 Dock5 后 YTHDF2水平下降,且可被蛋白酶体抑制剂 MG132 处理逆转。综上所述,Dock5 缺失促使YTHDF2蛋白水解,从而减缓 YTHDF2 介导的 m6A依赖性mRNA 衰减,上调了 LXRα 的表达。
图5 Dock5 通过 m6A 依赖性机制调节LXRα mRNA 的降解
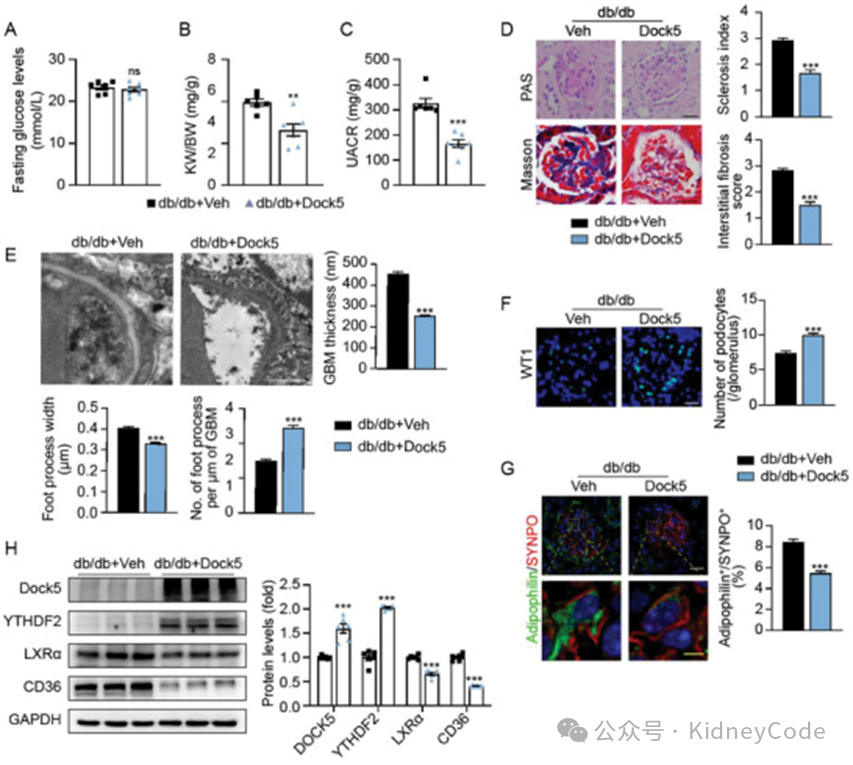
最后,作者构建了 Dock5 过表达小鼠,该小鼠在DKD 发病后 KW/BW 和 UACR 显著降低,足细胞足突增宽和消失、足细胞丢失和脂质积聚现象减轻。Dock5 过表达后,YTHDF2表达增加,LXRα 和 CD36 表达减少。这表明,改善Dock5 的降低可减轻蛋白尿性肾病的进展。(图6)
图6 Dock5 过表达可减轻 DKD 小鼠足细胞脂质积聚
文章结论与展望
综上所述,研究发现了足细胞中,Dock5 减缓YTHDF2 蛋白水解,从而加剧 YTHDF2 介导的 m6A 依赖性mRNA 衰减,下调了转录因子 LXRα 的水平,进而下调 CD36表达,从而改善足细胞对脂肪酸的摄取,降低足细胞脂毒性。
研究证明了 Dock5 在足细胞脂毒性和蛋白尿性肾病中的作用;发现了 CD36 可受 LXRα 调节;提出了 m6A可能在足细胞的脂肪酸代谢中发挥重要作用。发现CD36 抑制剂 SSO处理、LXRα 拮抗剂 GSK2033处理、过表达Dock5 等方式可显著减轻足细胞损伤,提示了 Dock5 可能成为蛋白尿性肾病的潜在治疗靶点。
来源:KidneyCode
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017