



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




点击图片,进入病例专栏↑↑
团队:孙贺 李玲
单位:中国医科大学附属盛京医院内分泌科
一个病理诊断为睾丸间质瘤的18岁男孩,在即将面临手术切除双侧睾丸之际,内分泌科会诊认为病因是先天性基因异常导致的肾上腺问题。
手术暂停。但随后的全外显子组基因检测结果未显示与疾病表型高度相关的致病性明确的基因变异。
“哪里出了问题呢?”内分泌科医生虽有疑惑,但未气馁,而是继续进行相关基因检测,最终证实了基因变异,并明确诊断,使得患者免于手术、依靠口服药治疗便成功缩小了肿大的睾丸。
患者男性,18岁,以“发现双侧睾丸肿大1个月”为主诉入院。
患者1个月前在他人提示下发现双侧睾丸肿大,双侧鸭蛋大小,无红肿疼痛。
2020年8月10日于当地医院行相关检查:超声检查提示双侧睾丸、附睾炎性改变不除外,双侧精索静脉曲张;泌尿系CT 检查提示双侧肾上腺改变;精液检查示精子数量为0;左睾丸肿物手术活检,结果提示性索间质肿瘤。
2020年9月2日于我院进行睾丸肿物活检组织病理会诊,结果为睾丸间质瘤。
2020年9月10日,患者为尽快行睾丸肿物切除手术,来我院泌尿外科,门诊以“睾丸肿物”为诊断收入院。
2020年9月15日,泌尿外科拟行睾丸肿物切除手术,术前请内分泌科会诊协作诊治。
患儿母亲共妊娠2次,第一胎不良妊娠结局,孕3个月时胎死宫内;该患儿为第二胎,于妊娠38周剖宫产,出生体质量3.5 kg,出生时皮肤偏黑,瘦小,未见外生殖器异常。
患儿出生3个月时食欲差,皮肤弹性差,无恶心呕吐,曾就诊当地医院儿科,诊断“胼胝体发育不良”和“酸碱不平衡”,具体化验检查未见。
8岁前饮食欠佳,纳差。8岁后开始出现第二性征表现,声音变粗、喉结显现、阴毛生长、阴茎发育较同龄儿大,身高增长加速,身材明显高于同龄儿。
15岁之后身高未再增长。16岁后身高低于同龄儿。有阴茎勃起,无遗精。
否认家族类似遗传疾病史。
血压130/80 mmHg,心率92 bpm,身高169.5 cm,体重65 kg,体质指数22.49 kg/m2,上部量86 cm,下部量83.5 cm,指间距170 cm。
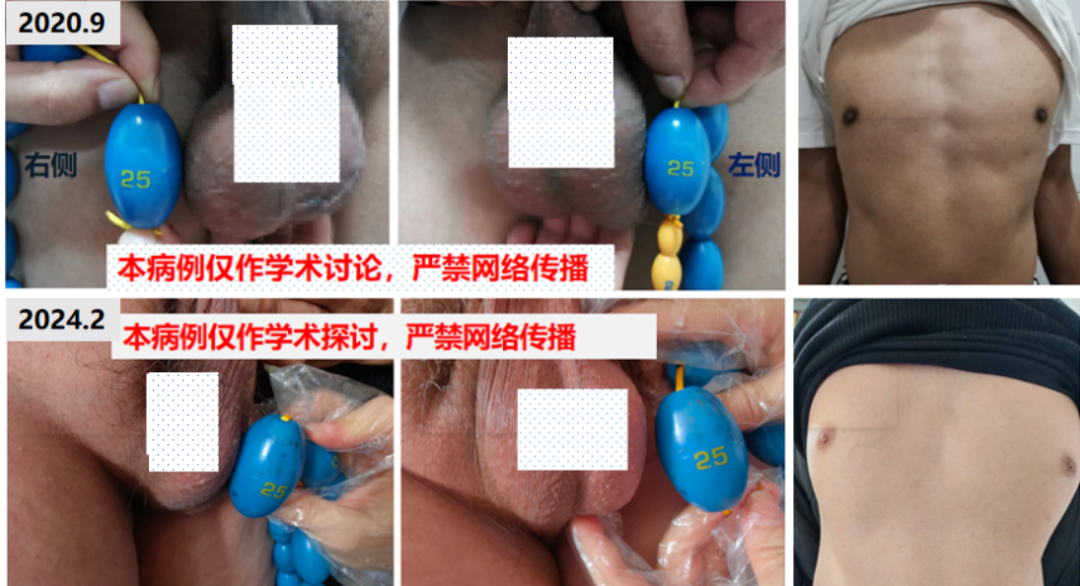
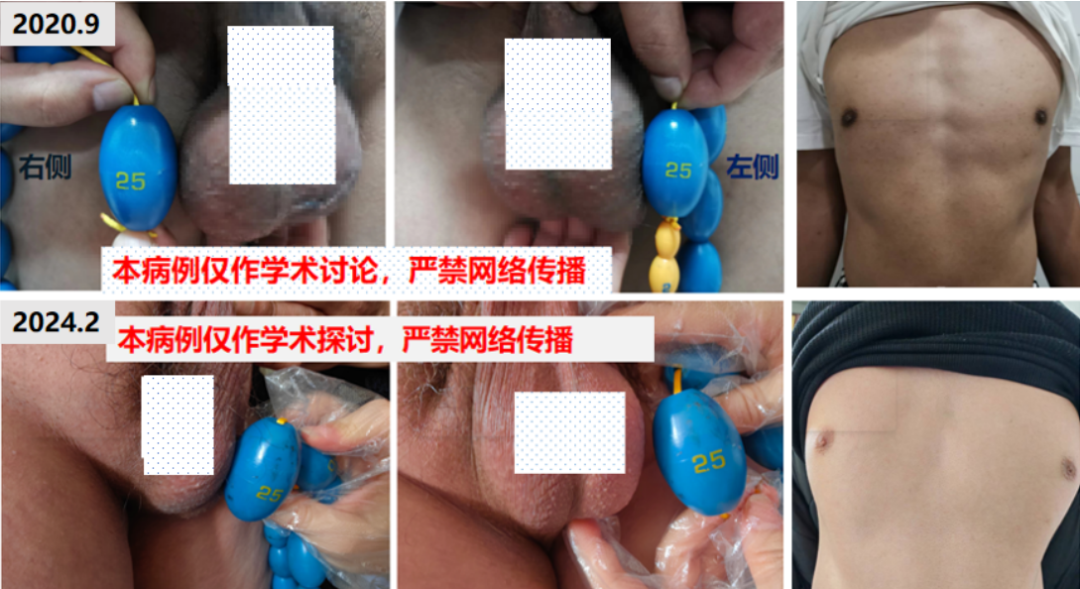
全身皮肤可见色素沉着,齿龈发黑,乳晕发黑,皮肤略粗糙,唇周无胡须,喉结明显,面部无痤疮,无腋毛,有腿毛。外生殖器TannerV期,阴毛呈男性分布。阴茎长约10 cm,双侧睾丸明显增大,右侧大小约 6 cm×4.5 cm×4 cm,左侧大小约6 cm×4 cm×4 cm,表面凹凸不平,质硬,无压痛。
性激素、肾上腺激素等相关检测(2020年9月10日):促卵泡生成素(FSH)0.25 mIU/ml;促黄体生成素(LH)0.30 mIU/ml;雌二醇(E2)55 pg/ml;泌乳素(PRL)26.35 ng/ml;孕酮(P)>40.00 ng/ml;睾酮(T)7.91 ng/ml;硫酸脱氢表雄酮(DHEAS)>1000.0 μg/dL;游离睾酮(F-T)52.12 pg/ml;雄烯二酮(A2)>10.0 ng/ml;游离雄激素指数(FAI%)102.80%;人绒毛膜促性腺激素(HCG)<0.5 mIU/ml;17α-羟孕酮(17α-OHP)>20 ng/ml。
(08:00)促肾上腺皮质激素(ACTH)375.4 pg/ml,皮质醇(COR)2.33 μg/dl。
肾素、血管紧张素、醛固酮、血糖、胰岛素、钾钠氯离子等未见明显异常。
生殖系统彩超(2020年9月12日):双侧阴囊内实性肿物,(左)5.8 cm×3.5 cm×2.8 cm,(右)5.9 cm×3.6 cm×2.5 cm。建议进一步除外恶性肿瘤(cancer)。
全腹增强CT检查(2020年9月18日):双侧睾丸占位;双肾上腺占位,建议进一步除外转移;腹膜后淋巴结稍大。
睾丸增强MR(2020年9月18日):双侧睾丸占位,恶性可能性大。
性索间质肿瘤,结合免疫组化,符合睾丸间质瘤(leydig细胞瘤)。
病理诊断是肿瘤诊断的“金标准”,是临床医师确定患者临床诊断、治疗方案和评估预后的重要依据。病理诊断为睾丸间质瘤,那么这个“睾丸肿瘤”是肯定需要手术切除的。
但是,反常升高的孕酮引起了内分泌科医生的注意。孕酮升高可提示怀孕,但是对于这个男孩而言,孕酮升高就非同寻常。睾丸间质瘤可伴随雌激素、雄激素升高,但是不会引起孕酮如此之高,并且随后化验的17α-OHP明显升高。
此外,从影像学检查来看,难道患者同时存在“睾丸”和“肾上腺”两个部位的恶性肿瘤?因此,仅有病理诊断是不够的,还需要结合患者详细的病史以及实验室检查等明确诊断。于是,我们以反常升高的孕酮和17α-OHP作为线索寻找答案。
在人体类固醇激素的合成途径中,有一类罕见病叫做先天性肾上腺皮质增生症(CAH),因参与皮质醇生物合成的蛋白质和酶缺陷所致。当21-羟化酶(21-OHD)或11β-羟化酶活性降低或丧失时,可导致孕酮和17α-OHP升高,雄性激素合成增多,皮质醇合成减少,ACTH分泌增加,刺激双侧肾上腺增生。两者均可表现为男性性早熟、女性男性化。
那么,该患者是21-OHD缺陷症?或是11β-羟化酶缺陷症?下一步需要基因检测确诊。
除了上述激素变化之外,本例患者的另一特征性改变是“双侧睾丸肿瘤”。CAH患者可伴发性腺肿瘤,对于男性患者,双侧睾丸是最常见的肿瘤发生部位,被称为睾丸肾上腺残余肿瘤(TART)。TART属于良性病变,并非真正意义上的“恶性肿瘤”。所以,TART与睾丸间质细胞瘤组织病理学鉴别诊断非常困难,确诊需要基因检测。
全外显子组基因检测(二代测序)结果却未显示与疾病表型高度相关的致病性明确的变异。于是,我们又针对CAH进行了相关基因检测[测序联合多重连接探针扩增(MLPA)]。
测序报告CYP21A2基因呈杂合突变;CYP21A2基因c.955C>T(p.Gln319Ter)杂合性变异,变异类型无义突变,致病性评级为“Pathogenic”,遗传自母亲;CYP21A2基因c.1069C>T(p.Arg357Trp)杂合性突变,突变类型为错义突变,致病性评级为“Pathogenic”,遗传自父亲。患者父母临床表型正常,父母CYP21A2基因均呈单一位点杂合突变。
本例患者的突变为复合杂合子,并非纯合子。因此,确诊为CAH,21-OHD缺乏症伴TART。
本病治疗方案为长期口服皮质激素替代治疗,既补充肾上腺皮质醇分泌不足,又可抑制ACTH的过量产生,抑制肾上腺增生。临床上多选用氢化可的松和地塞米松。儿童、青春期前多选用氢化可的松。成年后也有文献报道,地塞米松对睾丸功能改善有积极意义。
2020年9月18日,初始给予地塞米松0.75 mg(1片)每天睡前口服。随访3.5年,治疗期间多次复查实验室检查(结果详见下表)。
超声复查:双侧睾丸弥漫性病变较前缩小。
2020年10月30日:(左)5.0 cm×3.0 cm×1.8 cm,(右)4.6 cm×2.7 cm×2.0 cm;
2020年11月30日:(左)4.9 cm×2.8 cm×1.5 cm ,(右)4.4 cm×2.7 cm×1.6 cm;
2021年3月11日:(左)4.0 cm×2.3 cm×2.1 cm,(右)4.0 cm×2.4 cm×1.9 cm;
2024年2月5日:(左)5.2 cm×2.6 cm×2.1 cm,(右)4.4 cm×2.6 cm×1.9 cm。
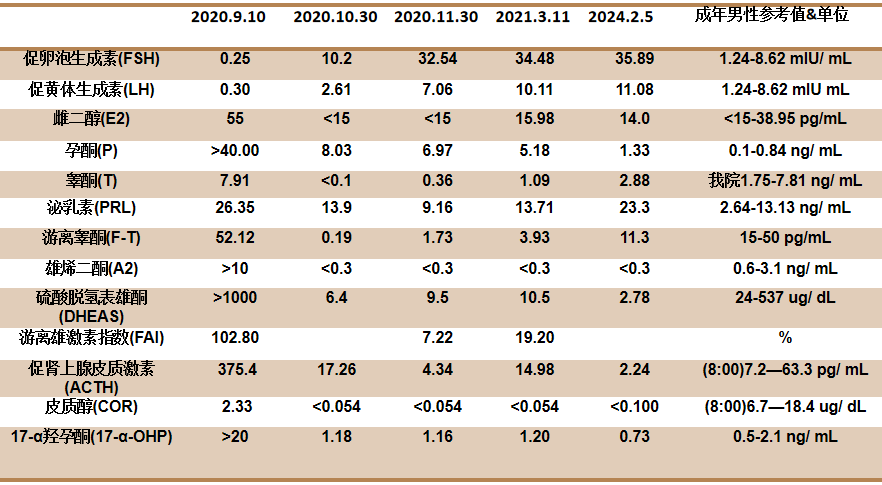
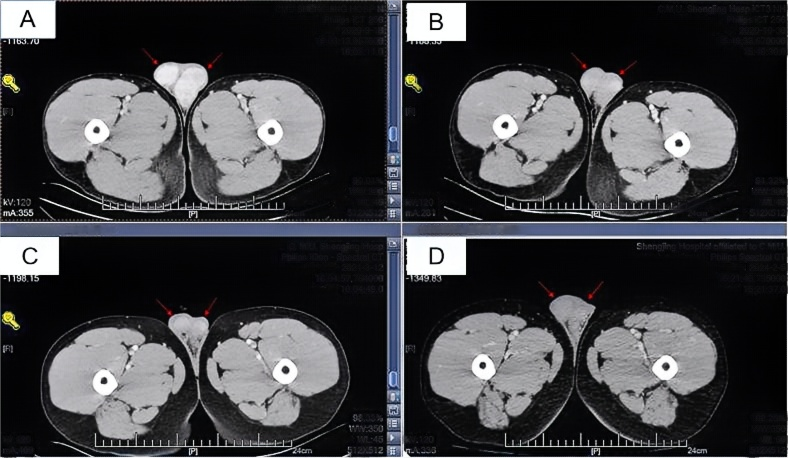
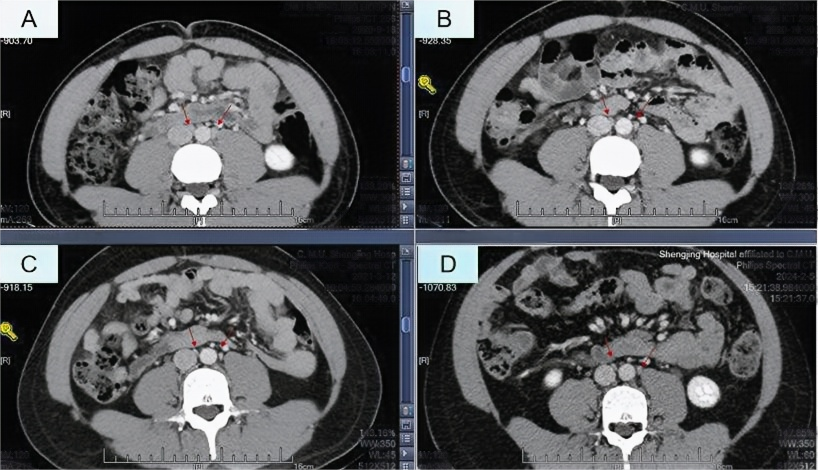
全腹增强CT检查复查:双肾上腺占位较前减小,肾上腺瘤瘤体较前缩小(图1);双侧睾丸占位,双侧睾丸体积较前减小(图2);腹膜后腹主动脉周围见稍大淋巴结较前减小(图3)。
图1 全腹增强CT检查:双肾上腺占位较前减小,肾上腺瘤瘤体较前缩小(A示2020年9月18日;B示2020年10月30日;C示2021年3月12日;D示2024年2月5日)
图2 全腹增强CT检查:双侧睾丸占位,双侧睾丸体积较前减小(A示2020年9月18日;B示2020年10月30日;C示2021年3月12日;D示2024年2月5日)
图3 全腹增强CT检查:腹膜后腹主动脉周围见稍大淋巴结较前减小(A示2020年9月18日;B示2020年10月30日;C示2021年3月12日;D示2024年2月5日)
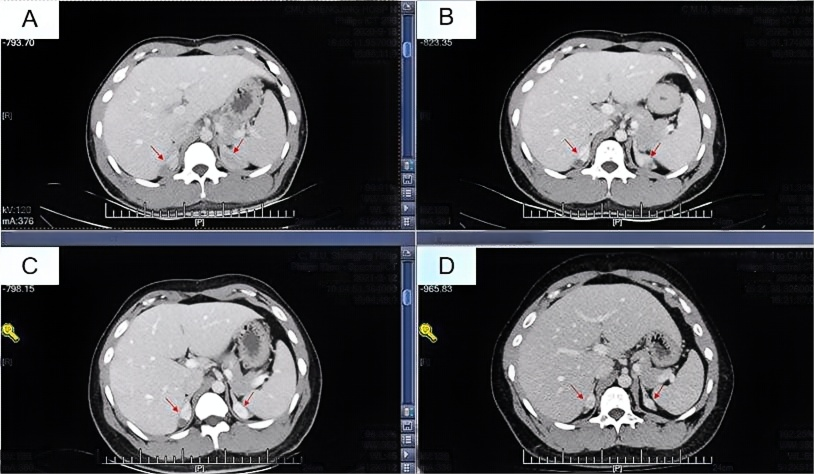
根据复查结果地塞米松剂量逐渐从1片减量至1/4片,治疗后患者自觉恢复良好,睾丸肿物较治疗前明显缩小,皮肤较前明显变白(图4)。
图4 治疗后患者自觉恢复良好,睾丸肿物较治疗前明显缩小,皮肤较前明显变白

遗憾的是,经过6个月、9个月乃至3.5年治疗后,精液复查显示精子数量仍然为0。
尽管TART为良性,极少恶变,但其增大的瘤体长期压迫曲细精管和损害睾丸周围环境会导致无精症或者少精症。小剂量激素的替代治疗,可免于手术切除,极大提高了患者的生活质量和心理健康。
因此,提高临床医生对此病认识,可以尽早发现和控制TART进展,使睾丸功能得到最大程度保护,也可减少误诊,以避免不必要的创伤性检查或手术。
CAH是一组少见的常染色体隐性遗传性疾病,是由于类固醇激素合成过程中某种酶先天性活性缺陷,导致相关激素的生成障碍。
21-OHD缺陷症是CAH中最常见的类型,占全部病例的90%~95%,主要病因是CYP21A2基因突变,导致21-OHD活性受损。
根据21-OHD的残余活性,可以将其分为两类
(1)经典型21-OHD缺乏症
失盐型(约75%):发病率1/20000,21-OHD活性0%,临床表现为呕吐、腹泻、脱水等,还会出现低血钠、高血钾、代谢性酸中毒等;
单纯男性化型(约25%):发病率1/60000,21-OHD活性1%,呈明显雄激素过多的表现;
(2)非经典型21-OHD缺乏症
患病率较高,为1/1000~1/2000,21-OHD活性为20%~50%,其症状隐匿不易诊断,或因高雄激素、卵巢多囊样改变和不孕等相似的临床表现易被误诊为多囊卵巢综合征(PCOS)。
TART发病率各地调查结果差异较大。在国外,CAH成人中TART发生率为14.3%~94%,儿童约24%,最小检出年龄2岁。在国内,儿童、青春期TART检出率为29.5%。研究发现,TART检出率与年龄呈正相关,15岁以后检出率明显升高,可达75%~100%。
TART最突出表现为睾丸肿物,大多数为双侧(75%~87%)、多灶性(16%)。
TART患儿基本无临床症状,早期的病变体检难以被识别。成年患者则多因不育或因检查发现少精、无精症时进一步检查才发现。
对于睾丸功能障碍,患儿睾丸功能受损可从儿童期即已经开始;进入青春期后,下丘脑—垂体性腺轴活性持续被压制,加重睾丸功能障碍状态;青春期后期,精液分析可帮助了解生育力。
75%的TART患者在经过充分糖皮质激素治疗后,瘤体会缩小、消退。对于大剂量糖皮质激素治疗仍无反应的患者,建议手术治疗。而早发现、早诊断、早控制对于最大限度保护患儿远期睾丸功能具有重要意义。
TART需要与非肿瘤性或肿瘤性睾丸肿物鉴别
非肿瘤性病变如睾丸结核,通过相关病史及症状,较易与TART区分。
肿瘤性病变包括生殖细胞瘤(精原细胞瘤、卵黄囊瘤、畸胎瘤),非生殖细胞瘤(支持细胞瘤、间质细胞瘤),睾丸淋巴瘤,睾丸白血病等。生殖细胞瘤具有特征性超声表现,且甲胎蛋白(AFP)、人绒毛膜促性腺激素(HCG)等肿瘤标志物升高。TART与睾丸间质细胞瘤和支持细胞瘤鉴别较难,尤其是睾丸间质细胞瘤,临床最易混淆。
鉴别要点
病史:TART只见于CAH患者,而睾丸肿瘤一般无CAH病史。
好发人群:TART多为青春期和成年后,而睾丸肿瘤则多见于儿童和成人。
病变部位:TART 大多为双侧病变,而睾丸肿瘤则多为单侧。
实验室检查:TART患者血睾酮、雄烯二酮、硫酸脱氢表雄酮、孕酮、17α-OHD均升高,而睾丸生殖细胞瘤、间质细胞瘤患者血睾酮等雄激素指标也增高,但孕酮、17α-OHD一般不升高,且生殖细胞瘤可有血AFP、HCG 等增高。
糖皮质激素治疗:TART患者对糖皮质激素敏感,治疗后可有效缩小瘤体,而睾丸肿瘤则无明显改变。
肿瘤性质:TART为良性肿瘤,而间质细胞瘤中10%为恶性。
病理:TART与间质细胞瘤组织病理学鉴别诊断困难,组织学上残余瘤细胞类似于睾丸间质细胞和肾上腺皮质细胞。有研究报道TART的GATA3、GATA6等特异基因表达较高,一般无Reinke晶体及胰岛素样因子3(INSL3),雄激素受体阴性反应,有脂肪化生、广泛纤维化,核多形性、淋巴样聚集体、脂色素较常见;而间质细胞瘤可见到Reinke晶体及INSL3,雄激素受体阳性反应,无脂肪化生、广泛纤维化。需要注意的是,有研究报道CAH患者可同时并发间质细胞瘤病变,表明两种不同类型的肿瘤有共存的可能。
基因检测有助于明确诊断
根据生化检查,结合临床表现,可获得指向性的21-OHD诊断;基因检测是21-OHD确诊的“金标准”。
高通量测序(即二代测序)技术是CAH分子诊断的重要方法,包括靶向测序,全外显子组测序及全基因组测序等。
然而,对于CYP21A2基因而言,其存在同源度极高的假基因(CYP21A1P),这使得在分析数据时无法区分短片段的来源,导致二代测序结果不可靠。尽管如此,二代测序技术在非21-OHD型CAH病例的鉴别诊断中仍具有重要作用。
因此,检测CYP21A2 基因变异时,推荐采用特异性扩增结合Sanger测序以及MLPA,对整个CYP21A2基因进行全面的变异检测。当患者检测出基因变异时,需要对其父母进行溯源,以明确致病变异是否分布在两条等位基因上。
复合杂合子是指同一个基因的双等位突变。同一个基因在每条染色体均存在变异,但发生的位置不同。复合杂合子重要意义在于:两对及以上的等位基因可以在各自杂合状态下(同时也是隐性遗传),联合起来导致疾病(无需纯合突变致病,但造成与纯合突变相似的破坏性)。此种遗传病,父母双方均为致病基因携带者,他们每次生育孩子都有25%的可能性成为患者。
综上,TART虽为良性病变,但患者不仅面临CAH相关内分泌紊乱,还面临不孕不育的风险,给患者的生活质量造成极大不良影响。因此,提高临床医生对此病认识,早期诊断和治疗可避免不可逆的睾丸功能损害。
面对泌尿外科即将手术切除睾丸的18岁患者,内分泌科医生勇于挑战睾丸组织病理诊断和二代测序的阴性结果,依然以性激素产生路径中各个代谢产物水平异常为线索,坚信自己的临床诊断,并改变基因检测方法,最终确认了诊断,使得患者免于手术、依靠口服糖皮质激素治疗缩小了明显肿大的睾丸。
本例患者幼时食欲差,皮肤黑,有酸碱失衡,疑似为CAH失盐型。8岁时出现第二性征发育,表现为外周性性早熟,作为鉴别诊断也应考虑到CAH。
遗憾的是,该患者由于未及时就诊而错失了早期诊断的机会。直到18岁因双侧睾丸占位就诊,疑似睾丸肿瘤,检查发现肾上腺也有占位,这种情况下应首先检测性激素、肾上腺激素和肿瘤标记物等。
TART是发生在睾丸内的瘤样增生,其增生组织在形态学和功能均与肾上腺相类似。对于病理学疑难病例、罕见病例等,诊断出现分歧是正常的,临床上诊断还需要结合病史、实验室检查,以及影像学检查提示是否为双侧性、皮质类固醇治疗反应等,确诊需要基因检测。
本例患者基因诊断明确,发现与临床表型高度相关的CYP21A2基因存在两个致病性变异,构成了复合杂合变异。家系分析发现,这两个变异分别遗传自父母双方,也就是父母为表型正常的携带者,父母再孕的再发风险为25%,可进行三代试管助孕或于孕中期行产前诊断。
TART是CAH的不常见并发症,主要见于CAH长期控制不好的患者,由于睾丸内存在肾上腺残余细胞,在长期高ACTH水平的作用下可逐渐增生形成TART。
TART的主要治疗方案是糖皮质激素,效果不佳者可通过手术将残余瘤剔除。该患者在使用地塞米松治疗后取得了显著疗效,残余瘤明显缩小。因此,一旦确诊为CAH,建议男性定期行阴囊超声筛查,女性定期行卵巢及附件影像学检查。
临床上以发现睾丸或卵巢及附件处的肿瘤作为就诊原因的患者,应警惕CAH的可能;如果CAH患者睾丸或卵巢及附件处存在占位性病变,应考虑TART或卵巢肾上腺残余肿瘤(OART)的可能,并进行相关检查,避免误诊或漏诊。
临床医生应充分了解TART发病机制、临床诊疗及预后,认真加以鉴别,避免切除睾丸。规范合理的诊治和系统的长期随访对预后具有重要意义。

欢迎您参与“疑难病例诊疗提升”专栏互动!无论您想点评病例还是发表学习感悟,抑或对病例诊治仍有困惑,都可以扫描上方二维码给小编留言!
我们会定期精选并刊登读者互动内容,并提供精美小礼品以资鼓励,期待您的积极参与和分享,您的专业视角将为专栏增添更多学术价值与意义!


版权说明:本文系中国医学论坛报社内分泌学科编委会精心出品,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载请联系【中国医学论坛报今日内分泌】申请授权
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017