






200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过





点击图片,查看专区
撰稿:复旦大学附属华山医院 陈新雨
审核:复旦大学附属华山医院 奚剑英
人类基因组中包含超过100万个串联重复序列位点,由于其重复性,串联重复序列在基因组中有极高的突变率。基因编码区或非编码区发生串联重复序列异常扩增所导致的遗传性疾病被称为动态突变疾病,多累及神经肌肉系统。本次大会的研究报道涵盖眼咽远端肌病(OPDM)、眼咽肌营养不良(OPMD)、肌萎缩性侧索硬化(C9orf72-ALS)等其他动态突变疾病的致病机制、检测技术、治疗策略等方面的最新进展。
来自法国遗传与分子细胞生物学研究所的ManonBoivin教授报告了OPDM和OPML中GGC重复序列的致病机制:翻译成含有多聚甘氨酸(polyglycine,polyG)的毒性蛋白。通过开发针对不同蛋白的特异性抗体,团队在不同OPDM亚型患者肌肉细胞的核内包涵体中验证了polyG蛋白的存在。同时,团队构建了OPML、OPDM的LHCN-M2人骨骼肌细胞模型和AAV/MYO4A小鼠模型,在体内外证实了polyG蛋白聚集可在细胞质和细胞核内形成包涵体,并具有细胞毒性。此外,团队发现一种药物化合物,在小鼠模型中能够溶解polyG蛋白聚集体,恢复肌肉细胞活性,为OPDM的治疗提供了新的策略与希望。
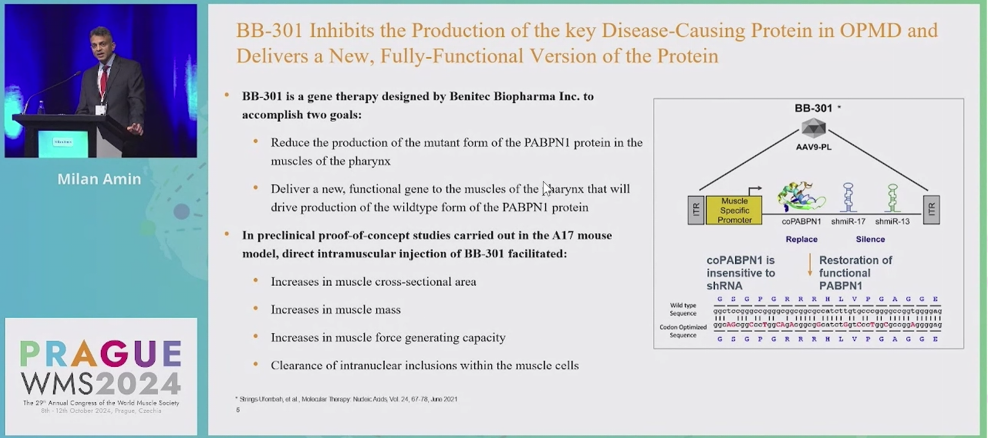
来自美国纽约大学Grossman医学院的Milan Amin教授介绍了OPMD最新的基因治疗方法——旨在改善OPMD患者的吞咽困难症状的BB-301的Ⅰb/Ⅱa期临床试验(NCT06185673)。BB-301使用新型改良的AAV9载体,同时表达经密码子优化的PABPN1蛋白和两个针对PABPN1突变体的siRNA,通过基因沉默抑制PABPN1突变蛋白表达,并允许正常PABPN1蛋白表达,维持正常功能。该临床试验的主要研究终点包括视频荧光吞咽检查(VFSS)对吞咽后总咽残余量(TPR)进行系列评估和患者报告的吞咽症状问卷(SSQ)。已有两名受试者接受了最低剂量的BB-301治疗,其中受试者1在第270天时SSQ和TPR指标均有改善,受试者2在第180天时SSQ改善幅度达到89%,已达到正常吞咽水平;在安全性方面,目前未报道重大不良反应。这些数据初步显示了BB301基因治疗成功改善OPMD患者吞咽功能的潜力。
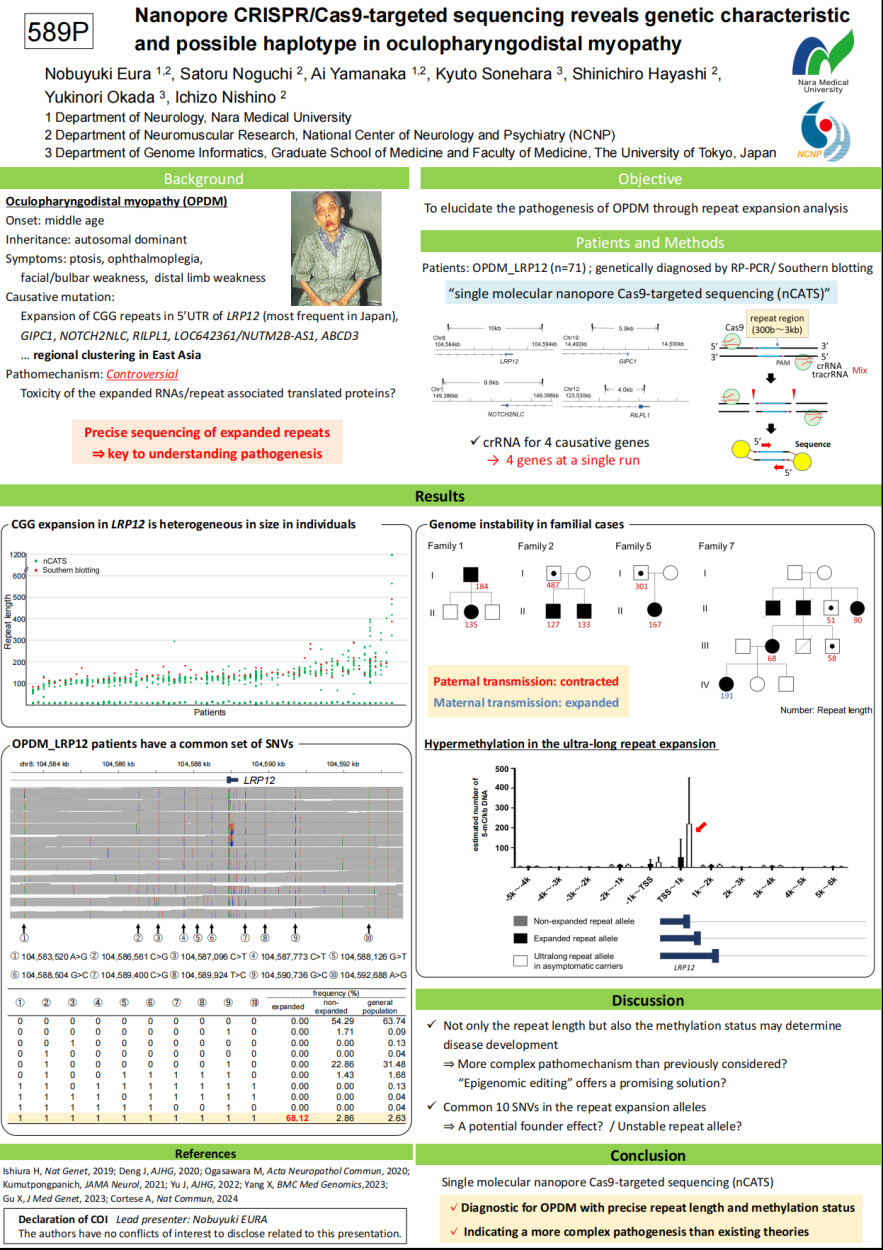
日本东京大学的研究团队使用纳米孔CRISPR/Cas9靶向测序(nCATS)技术精确描述了OPDM1的遗传特征,包括重复次数、甲基化状态和特定单倍型。通过对71名OPDM_LRP12患者的深入分析,研究团队发现:
①CGG重复扩增纯度与重复次数无关,且存在基因组不稳定性:重复次数在父系传递中缩减,而在母系传递中则扩增;
②在家族队列中,无症状个体的重复次数和甲基化水平高于有症状患者;
③在患者重复扩增的等位基因附近发现了一组独特的由10个单核苷酸变异(SNV)构成的单倍体型,携带该单倍型的正常个体也显示出轻微的CGG重复扩增(最长80bp),提示了LRP12基因5’UTR区CGG重复扩增可能存在奠基者效应(founder effect),特定单倍型可能与LRP12基因的不稳定重复相关。
上述发现加深了对OPDM的诊断和病理机制的理解。
法国索邦大学肌病学研究中心的研究团队采用了CRISPR/Cas9富集的纳米孔(ONT)长读长测序技术,对OPDM和DM1的基因进行了靶向精确分析,确定了三核苷酸重复次数、重复长度分布和DNA甲基化水平。通过不同的碱基调用策略(Guppy、Bonito和Dorado),成功区分正常和扩增的等位基因,强调了碱基调用策略在准确描述重复区域中的重要性,并提出了针对重复扩增疾病的CRISPR/Cas9富集长读长测序策略的指南,有助疾病的诊断。
日本东京国立神经精神研究中心的研究团队利用通过单核RNA测序技术(snRNA-seq)和免疫组织化学染色分析,在OPDM中可视化了两种不同类型的致病肌纤维,并将其与两条不同的转录组途径,神经源性(COL19A1)和肌源性途径(NCAM1)相关联,为研究肌肉病理作为慢性肌病的生物学标志物提供了新思路。
法国肌病学研究中心的研究团队通过对CLIP-seq数据库的生信分析,筛选出RNA结合蛋白(RBP)FUS/TLS作为PABPN1表达的潜在调节因子。通过siRNA和过表达系统,发现FUS/TLS的缺失影响了PABPN1在RNA和蛋白水平上的调控变化。团队正开展进一步的下游分析,包括力学测量、组织学和分子分析,提示FUS/TLS可能是OPMD疾病进程及肌肉老化过程中调控PABPN1表达的潜在分子靶点。
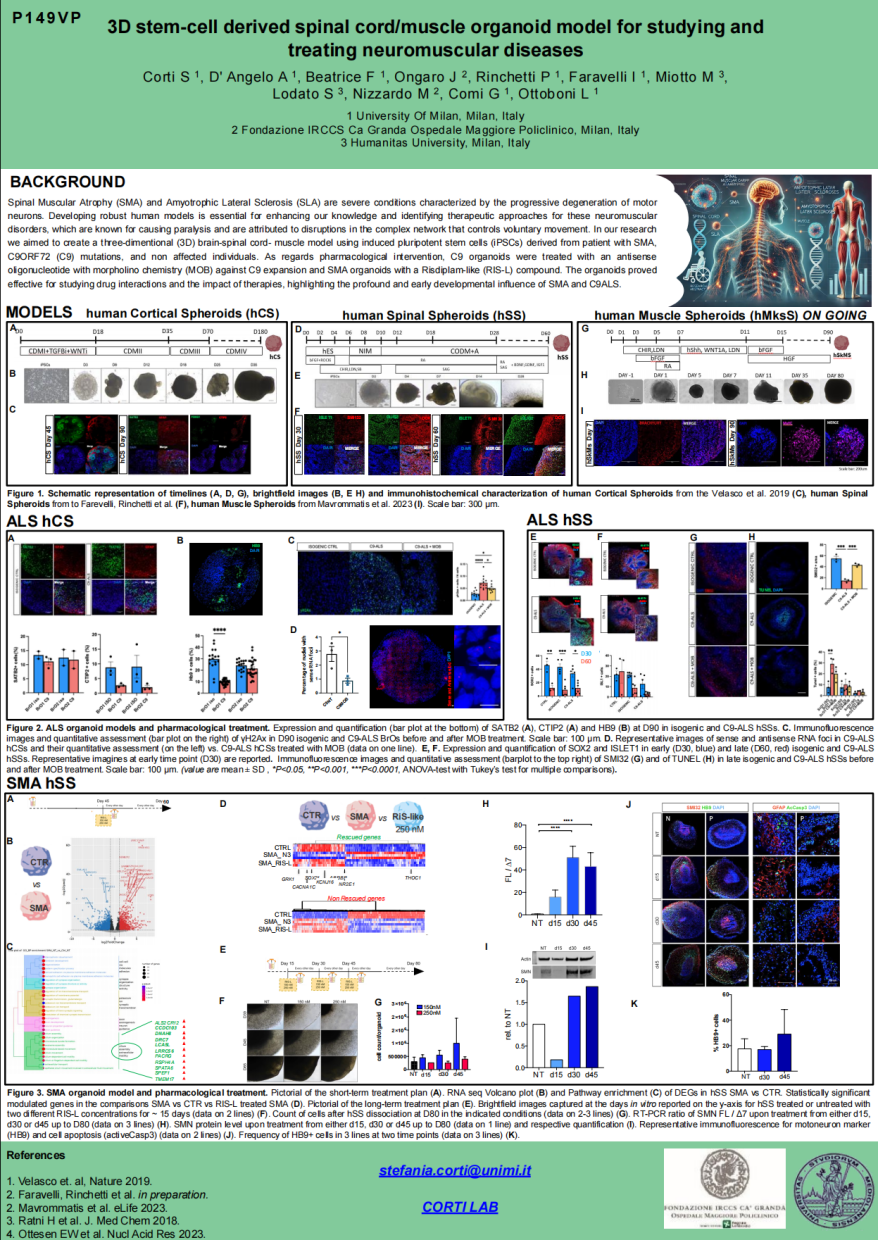
意大利米兰大学的研究团队利用脊髓性肌萎缩症(SMA)和肌萎缩性侧索硬化症(C9orf72-ALS)患者来源的诱导多能干细胞(iPSCs)开发了脊髓和骨骼肌类器官3D模型用于神经肌肉疾病的研究。通过全面的转录组、单细胞测序、免疫表型分析等方法,显示这些疾病具有发育相关的特性。在药理学方面,使用了Risdiplam样化合物(RIS-L)和针对C9orf72相关ALS的反义寡核苷酸(MOB)进行干预。对于SMA类器官,RIS-L化合物有效纠正了全长SMN2蛋白与截短SMN2蛋白的比例,并显示出显著的体外持久性;对于ALS类器官,MOB有效减少了DNA损伤和毒性RNA聚集。这些模型为研究药物相互作用以及治疗效果提供了有效的体外工具,同时也强调了SMA和ALS对发育早期的深远影响。
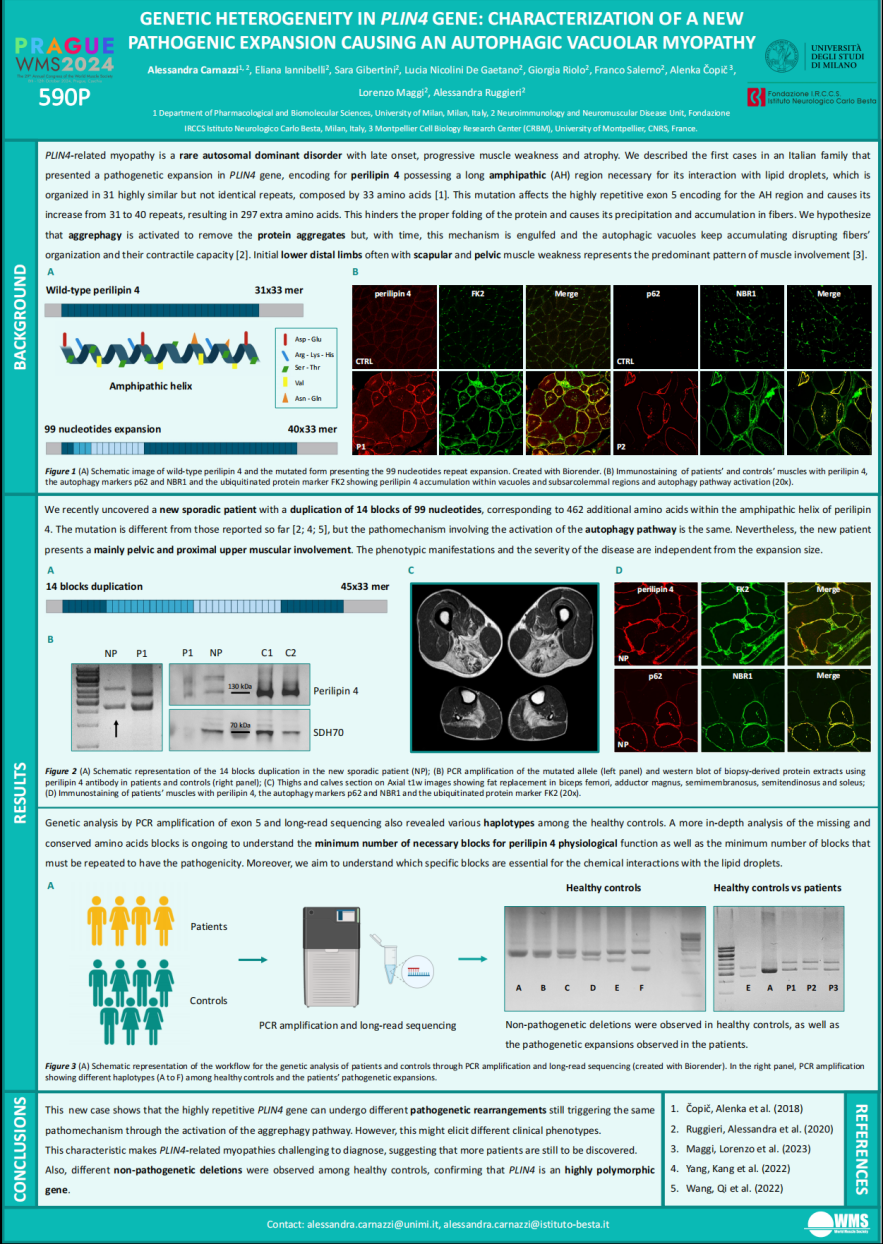
PLIN4相关肌病是一种罕见的远端肌病,该致病突变影响编码的脂包蛋白4(Perilipin4),其两亲性(AH)区域由31个高度相似的重复序列组成,每个重复序列由33个氨基酸组成。研究团队既往报道了首例意大利PLIN4相关肌病,其AH区域的重复扩增为40个,产生了297个额外氨基酸。团队最近又报道了一个新的散发病例,携带14个99次的核苷酸重复,导致462个额外氨基酸,该患者表现为近端的肌肉受累模式,蛋白聚集物的积累触发了自噬途径的激活。PLIN4基因重复扩增的复杂性使得其相关肌病的基因筛查具有很高的挑战性;团队推测有更多的病例尚未被发现,疾病的致病机制仍需进一步阐明。
原创内容,转载需授权!
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017