


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




先天性肌无力综合征(Congenital Myasthenic Syndromes, CMS)是一组遗传性、可治疗的神经肌肉传递障碍疾病,导致肌肉无力和易疲劳,目前已有35个相关基因被报道。CMS通常在出生或婴儿期发病,但也有部分患者在儿童晚期或成年期出现症状。CMS的发病率因地区而异,均为罕见病。据不完全统计的患病率,英国每百万人中约2.8–14.8人,巴西每百万人中约1.8人,斯洛文尼亚每百万人中约22.2人。
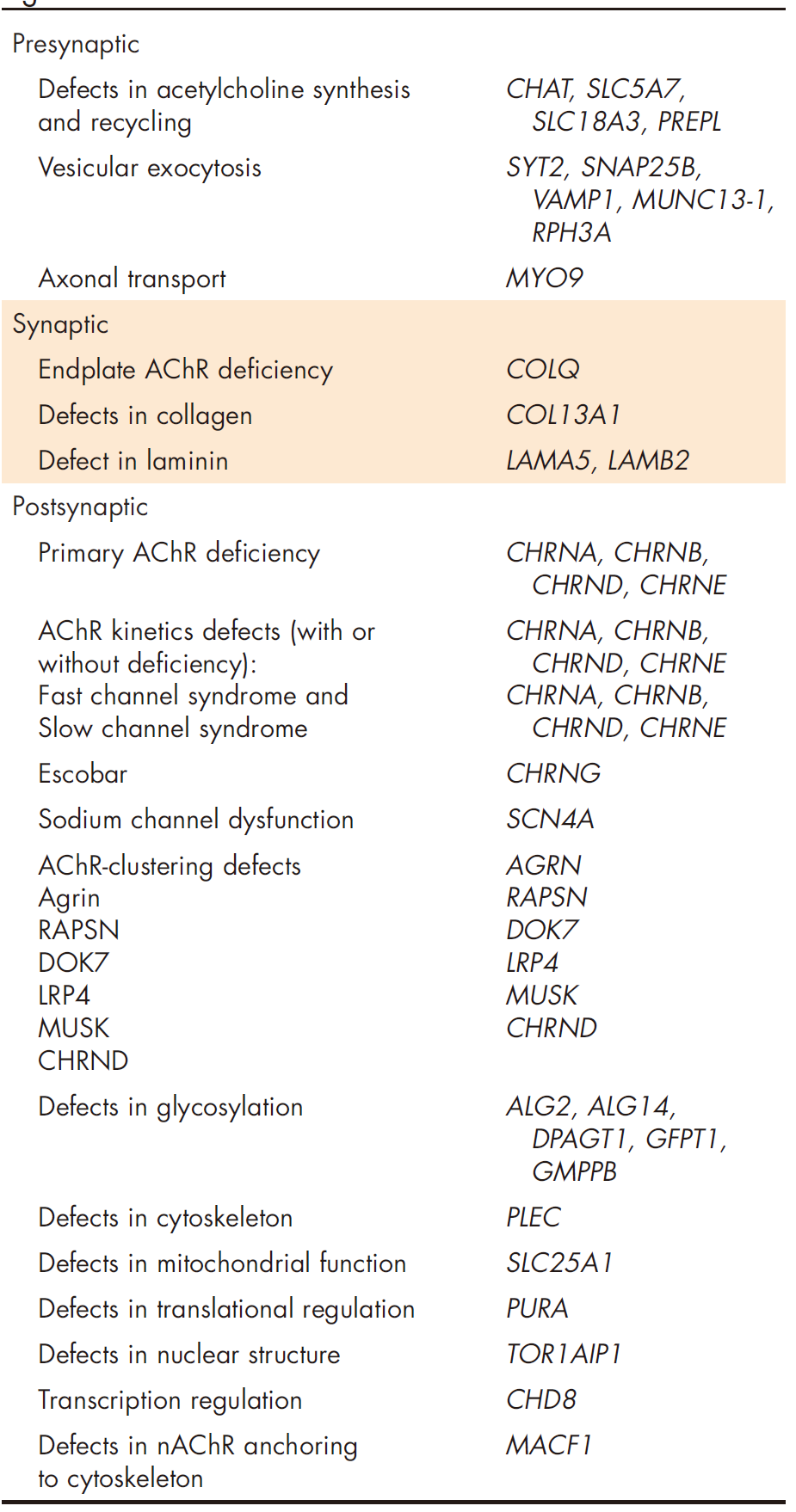
CMS根据其病理机制和分子靶点被细分为多个类别。目前已发现的35个相关基因分类如下:
1.突触后CMS:这一类涉及肌肉乙酰胆碱受体(AChR)及其相关蛋白的基因突变,如CHRNA1、CHRNB1、CHRND和CHRNG。突触后CMS占据大多数,主要特征是AChR数量减少或功能异常,导致肌肉传导受阻。
2.突触前CMS:涉及运动神经末梢的蛋白编码基因突变,如RPH3A和CHAT等。这些基因影响乙酰胆碱的合成、释放及囊泡稳定性,进而影响神经肌肉接头的传递效率。
3.突触CMS:包括COLQ、LAMB2和LAMA5等基因,这些基因编码的蛋白在基底膜的形成和维持中起关键作用。突变会破坏神经肌肉接头的结构完整性,影响肌肉收缩功能。
4.跨膜蛋白和信号转导相关CMS:如LRP4、MUSK和DOK7,这些基因参与神经肌肉接头的结构维护和信号转导。突变会导致AChR簇集和稳定性下降,影响神经信号的有效传递。
5.糖基化缺陷型CMS:如GFPT1、DPAGT1、ALG2、ALG14、GMPPB等基因突变:影响蛋白质的糖基化过程,导致NMJ相关蛋白质功能紊乱,影响神经肌肉传导的稳定性和效率。
6.非神经肌肉接头相关CMS:一些基因如PURNA和TOR1AIP1虽然在神经肌肉接头中发挥作用,但其广泛表达会导致患者表现出多系统受累的临床特征,影响范围超出单一神经肌肉接头。
通过对CMS的具体分类,可以更精准地理解其发病机制,为临床诊断和治疗提供依据,推动个性化医疗的发展。
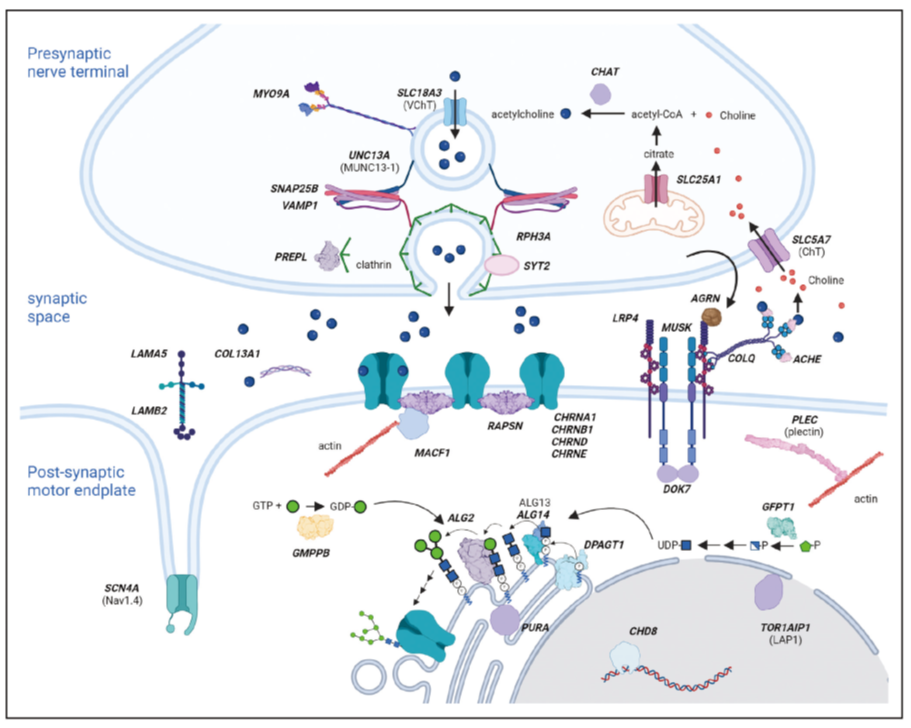
图1. 展示了与不同先天性肌无力综合征相关的基因产物如何参与神经肌肉接头生物学的过程。基因名称以加粗斜体大写字母表示。如果蛋白质名称与基因名称不同,则在基因名称下方括号中标注蛋白质名称。
该综合征主要通过常染色体隐性遗传和常染色体显性遗传两种方式传递,表现出不同的遗传模式和临床特征。
在常染色体隐性遗传的CMS亚型中,大多数病例需要患者从每位父母处各继承一个突变等位基因才能发病。这类亚型通常涉及双等位基因的基因突变,常见的相关基因包括AGRN、LRP4、MUSK、COLQ、GFPT1、DPAGT1、PLEC1、LAMB2、LAMA5及COL13A1等。这些基因突变主要影响神经肌肉接头的结构和功能,降低神经信号传递的效率,导致肌无力及其他相关症状。
部分CMS亚型则遵循常染色体显性遗传模式,仅需一个突变等位基因即可引发疾病。显性遗传的CMS包括慢通道综合征、SNAP25突变相关的CMS、PURA突变相关的CMS及部分SYT2突变相关的CMS等。慢通道综合征由于乙酰胆碱受体通道的异常持续开放,导致肌肉疲劳;SNAP25和SYT2突变则影响神经递质的释放机制;PURA突变不仅影响神经肌肉接头,还涉及中枢神经系统功能,可引发多系统症状。
CMS的发病机制复杂多样,主要由多种基因突变引发不同的分子异常。具体机制包括:某些基因突变减少了肌肉膜上乙酰胆碱受体(AChR)的数量,降低了对乙酰胆碱的响应能力;AChR的离子通道功能受损,导致电流传导异常,影响肌肉收缩能力。此外,MUSK和LRP4等基因的突变干扰了AChR的簇集和稳定性,使其无法在神经肌肉接头处稳定存在。
乙酰胆碱的合成与释放亦可能因基因突变而受阻,削弱了神经肌肉传递的效率。基底膜结构的异常,如COLQ和LAMB2基因的突变,破坏了神经肌肉接头的稳定性,进一步影响信号传导。此外,PURNA和TOR1AIP1等基因的突变不仅影响神经肌肉接头,还波及其他系统功能,导致多系统症状的出现。因此,CMS通过多种基因突变和分子机制,影响神经肌肉传导,导致临床上表现出多样化的肌无力症状。
先天性肌肉无力综合征(CMS)的临床表现多样,主要特征包括肌无力和易疲劳,尤其在重复肌肉运动后症状加重。此外,患者常见面部及眼肌无力,如眼睑下垂和眼球运动受限。严重病例可能出现呼吸肌无力,导致呼吸困难甚至需机械通气支持。患者可表现为步态异常和四肢近端肌肉无力,影响日常活动能力。
某些CMS还可能伴有肌张力下降,导致婴幼儿期肌张力低下。非神经肌肉接头相关的CMS患者可能出现多系统症状,如发育迟缓、癫痫、肾病及面部抽搐等。症状多在出生或婴儿期发病,但部分病例也可能在儿童晚期或成年期出现。
COLQ基因突变相关的CMS不仅导致全身性肌无力和呼吸困难,部分患者还可能出现眼睑下垂及骨骼畸形。LAMB2和LAMA5基因的突变分别与Pierson综合征和面部抽搐等多系统表现相关,影响肾脏、眼部及神经系统。PURA和TOR1AIP1基因突变的CMS患者表现出发育迟缓、癫痫及特定的肌无力症状。PLEC1基因突变则导致皮肤和肌肉系统的双重损害,如表皮大疱性疾病和肢带型肌营养不良。
SCN4A基因突变相关的CMS不仅引起肌无力和肌痛,还可能导致周期性麻痹和肌张力异常。涉及糖基化缺陷的CMS,包括GFPT1、DPAGT1等基因突变,主要表现为肢带型肌无力,可能伴有认知障碍及其他器官系统的影响。COL13A1及其他基因如AGRN、LRP4等突变的CMS亚型,虽主要表现为肌无力,但也可涉及呼吸系统、认知功能及骨骼异常等多系统。
诊断CMS依赖于临床表现、家族史、实验室检查及遗传检测,主要步骤包括:通过详细病史采集和体格检查,重点评估肌力减弱的部位、症状发作时间及家族遗传背景。进行电生理检查,如重复神经刺激(RNS)和单纤维肌电图(SFEMG),以检测神经肌肉传递功能和同步性。通过生化检查评估患者对乙酰胆碱酯酶抑制剂的反应以及血清和肌肉组织的代谢指标,帮助分类不同类型的CMS。
在基因检测方面,采用基因panel或全基因组测序技术,识别已知或新型病因突变,确诊疾病类型。在必要时,进行肌肉和神经组织活检分析病理变化,辅以影像学检查如MRI,以排除其他神经肌肉疾病。这一综合诊断流程确保了CMS的准确识别和分类,为后续治疗提供依据。
在临床实践中,CMS与肌营养不良症在症状上存在显著重叠,导致部分CMS亚型容易被误诊为肌营养不良症。
糖基化缺陷相关的CMS因涉及多个基因突变(如GFPT1、DPAGT1、ALG2、ALG14、GMPPB)而表现出肢带型肌无力、肌酸激酶(CK)升高及肌肉活检异常等特征,与肢带型肌营养不良高度相似。PLEC1突变相关CMS亦表现出表皮性大疱性疾病和肢带型肌营养不良,容易产混淆。COL13A1突变相关的CMS尽管主要影响神经肌肉接头,但其早发性近端肢体肌无力、脊柱畸形及认知障碍等临床表现,与某些早发型肌营养不良症存在相似之处,容易导致误诊。
误诊的主要原因在于CMS与肌营养不良症在肌无力分布、发病时间、CK水平及肌肉活检结果上可能存在相似性。为避免误诊,临床应详细采集病史和家族史,进行电生理检查(如重复神经刺激和单纤维肌电图),并采用基因测序技术进行精准诊断。此外,多学科团队的综合评估也是确保正确诊断的重要手段。
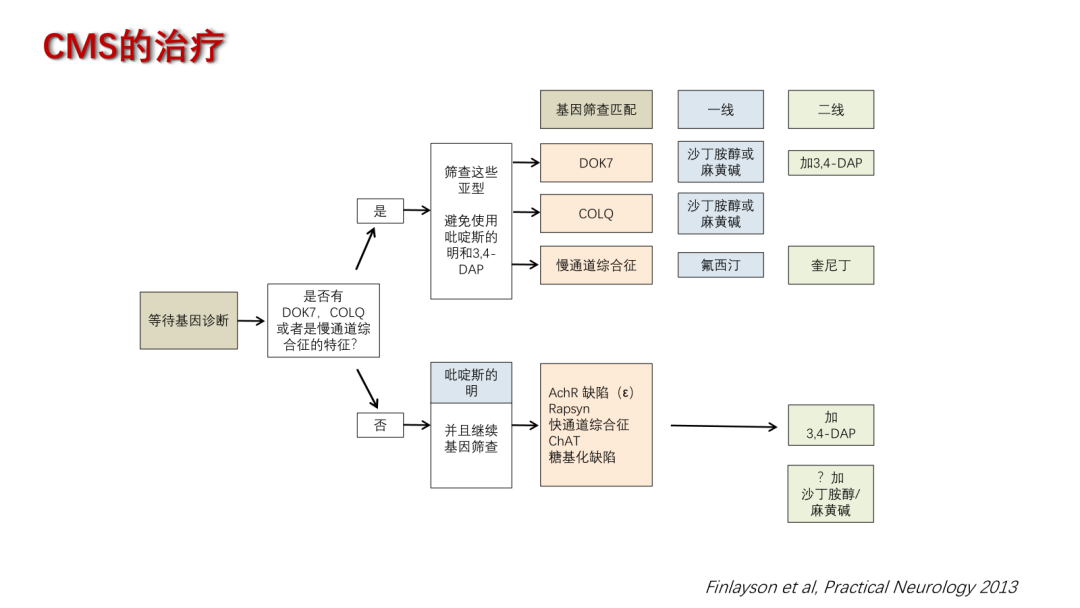
CMS虽为遗传性疾病,但多种治疗策略已被证实能有效改善患者症状。在药物治疗方面,乙酰胆碱酯酶抑制剂(如新斯的明或吡啶斯的明)通过增加神经肌肉接头处乙酰胆碱浓度以提升肌力,但DOK7、COLQ和慢通道综合征的肌无力却会加重。4-氨基吡啶用于增强神经肌肉传递,改善肌肉功能。沙丁胺醇作为一种β₂-肾上腺素受体激动剂,已被用于治疗某些类型的CMS,包括DOK7、COLQ、AChR和PURA等基因突变类型。
在基因治疗领域,腺相关病毒(AAV)介导的基因转导技术可针对特定基因突变(如CHAT CMS)恢复正常基因功能,CRISPR-Cas9等基因编辑技术则有潜力用于修复致病突变,但处于研究阶段。正向全osteric调节剂及氯离子通道抑制剂(如NMD670)能够增强受体功能和缓解肌肉症状。
支持治疗包括物理治疗和康复,旨在维持肌肉力量和功能、预防肌肉萎缩,在呼吸肌无力严重时提供机械通气支持。对于多系统症状的CMS,需综合管理如肾病、癫痫等非神经肌肉接头相关症状,采取多学科治疗策略。
尽管在CMS的理解与治疗方面取得了显著进展,但仍面临诸多挑战。CMS的病因具有高度多样性,目前已识别的基因突变仅涵盖部分病例,亟需进一步研究以揭示更多致病基因。临床表型的异质性使得同一基因突变可能导致不同的临床表现,增加了诊断和治疗的复杂性,这需要更多的病例研究和数据积累。
个体化治疗成为重要课题,不同类型的CMS对治疗反应各异,精准制定基于基因和分子机制的治疗方案至关重要。尽管基因治疗展现出巨大潜力,其长期安全性和有效性仍需大量临床试验证实。对于涉及多系统的CMS患者,如何协调多学科治疗以提升生活质量也是未来研究的重点。同时,随着对CMS病理机制的深入理解,开发新型治疗靶点和药物以提升治疗效果成为必然趋势。
综上所述,未来的研究应聚焦于基因多样性、个性化治疗、新疗法开发及综合管理,以应对CMS领域的挑战并推动其发展。

Congenital myasthenic syndromes:increasingly complex. Curr Opin Neurol. 2024 Oct 1;37(5):493-501.
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017