


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:北京协和医院 郭潇潇

郭潇潇 主治医师
本文要点提示:
21岁年轻女性,心脏扩大、全心衰、结性心律,心脏病变是原发?是继发?营养不良、生长发育迟缓、第二性征发育不良、面容异常、四肢骨骼异常,是偶然?是必然?这位年轻患者的“诡异”病情牵动着北京协和医院无数医护的心。在同情心的驱使下、在不轻言放弃的精神激励下,他们开始了艰难的探索。
病例简介
基本情况
患者,女性,21岁,因“头晕2年,活动后胸闷1年,加重1月”收入我科。2年前于剧烈活动时出现头晕,伴乏力、心悸,偶有黑矇,无视物旋转、意识丧失,休息数分钟可缓解,未就诊。1年前出现活动后(慢跑500米、上五层楼)胸闷、气短,休息10分钟左右可缓解。上述症状逐渐加重,至入院前1月爬2层楼气短明显并出现腹胀、恶心、纳差、少尿、双下肢可凹性水肿及夜间阵发性呼吸困难伴咳嗽、咳白色泡沫痰。就诊当地医院。给予利尿治疗后胸闷、气短有所缓解,双下肢水肿减轻,夜间可平卧入睡。发病以来,精神、睡眠差,食欲下降;二便基本正常。
辅助检查
心电图提示交界区逸搏心律(图1);胸腹CT提示双侧少量胸腔积液,少量腹水;超声心动图提示右心增大,右室及左室运动弥漫性减弱,左室射血分数(LVEF)35%,三尖瓣重度反流,二尖瓣中度反流。
既往史
12岁开始出现双手指尖遇冷变色(先变白后变紫),无明显疼痛;近半年出现脱发,否认皮疹、光过敏、关节肿痛,无口眼干、口腔外阴溃疡。
个人史
第二胎、第二产,足月顺产,头先露,无窒息,出生体重及身长正常。母亲40岁怀孕,孕期无疾病及用药史。母乳喂养至1岁,出牙、说话、走路与同龄人相仿。自3岁起较同龄人矮、瘦,无偏食,智力正常。
月经婚育史
15岁初潮,行经7天,月经周期30天;2年前开始月经紊乱,月经周期2~4月,行经3~7天。未婚。
家族史
否认家族类似病史。父亲患卒中,高血压;母亲及哥哥体健。父亲身高170 cm,哥哥身高170 cm,母亲身高165 cm。
入院查体
身高155 cm,体重30 kg,体质指数(BMI)12.49 kg/m2,心率52次/分,血压100/50 mmHg,体形消瘦,斜肩;四肢皮下脂肪明显减少;皮肤干燥;头发稀疏无光泽,小下颌,尖鼻,高颚弓,齿列拥挤;颈静脉充盈,胸腹壁浅静脉曲张,全身散在咖啡色素斑,无腋毛,阴毛Ⅲ级,乳房Ⅳ期。心尖搏动位于第5肋间左锁骨中线内1 cm,心律齐,P2=A2,心尖部吹风样收缩期2/6级杂音,向左腋下、左肩胛区传导;胸骨左缘4肋间可及3/6级全收缩期杂音,向心尖区传导。肺部查体未见明显异常;腹膨隆,无压痛,肝肋下4 cm,移动性浊音阴性;胸腰段脊柱略凸向左侧;四肢瘦,关节突出,双下肢轻度凹陷性水肿;四肢深浅感觉及肌力正常;扁平足,双侧外翻畸形。
入院后进一步检查
常规及免疫相关检查
便常规潜血(+)、肝肾功能未见异常;红细胞沉降率(ESR)、超敏C反应蛋白(hsCRP)、蛋白电泳、免疫球蛋白均正常范围。
内分泌及代谢相关检查
甲功、血乳酸均正常;总胆固醇(TC)3.38 mmol/L,甘油三酯(TG)1.11 mmol/L,低密度脂蛋白胆固醇(LDL-C)2.16 mmol/L,高密度脂蛋白胆固醇(HDL-C)0.84 mmol/L;糖耐量、生长激素、血促肾上腺皮质激素(ACTH)、24小时尿游离皮质醇、泌乳素正常;性激素检查示卵泡刺激素(FSH)51.9 mIU/ml,雌二醇(E2)31.4 pg/ml,促黄体生成素(LH)68.81 mIU/ml,孕酮(P)0.11 ng/ml(激素水平提示处于绝经期);血尿氨基酸筛查+肉碱谱未发现特异性氨基酸、脂肪酸代谢异常;
结缔组织病筛查
抗核抗体(ANA)、抗可提取核抗原(ENA)、抗中性粒细胞胞浆抗体(ANCA)、自身免疫性肝炎抗体、抗心磷脂抗体(ACL)、抗Jo-1、抗Scl-70均阴性。
影像学检查
胸片正侧位显示心影增大,胸廓呈梨形;腹部超声示肝大、剑突下2.7 cm、肋下3.0 cm,三支肝静脉增宽;泌尿系超声未见异常;妇科超声示子宫内膜偏薄。大血管超声(上下肢动静脉、颈动脉、椎动脉、锁骨下动脉)未见明显异常;肌电图未见异常;全脊柱相显示胸腰椎轻度左侧偏斜。
心脏相关检查
B型钠尿肽289 ng/L;肌钙蛋白I(cTnI)0.447 μg/L(<0.05 μg/L),余无异常;24小时动态心电图(Holter)示交界区心律,24小时总心搏数62063次,最快心率71 bpm,最慢心率36 bpm,平均心率43 bpm;超声心动图示心肌病变、右心增大、右室内可见较多调节束,左右室收缩功能减低、室壁运动普遍减低、LVEF39%、三尖瓣环收缩期运动幅度(TAPSE)12 mm,下腔静脉增宽(20 mm)、吸气变化率小于50%,重度三尖瓣关闭不全、中度二尖瓣关闭不全(图2)。心肌磁共振示右心明显增大,右室肌小梁增多,左右室壁收缩功能减弱,LVEF40.4%,右室射血分数(RVEF)49.5%;延迟强化示室间隔及左室前壁、前侧壁和下壁外膜下多发片状高信号(图3),考虑心肌病变,二尖瓣、三尖瓣、主动脉瓣关闭不全。
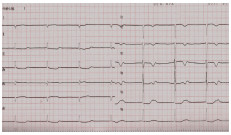
图1 心电图提示交界区心律,多导联ST-T改变

图2 超声心动图可见三尖瓣重度关闭不全

图3 心脏磁共振提示右心明显增大,右室肌小梁增多(A,B),室间隔及左室前壁、前侧壁和下壁心外膜下多发延迟强化
初探 本例为青年女性,慢性病程,多系统受累。心脏方面表现为右心增大,左、右室收缩功能减低的心肌病变伴持续交界区心律;幼年起生长发育迟缓,第二性征发育不良伴卵巢早衰;面容异常,包括小下颌、齿列拥挤、高颚弓、尖鼻;四肢骨骼异常可见斜肩、梨状胸、脊柱侧弯、四肢皮下脂肪明显减少、雷诺现象;皮肤干燥、脱发、散在色素沉着。
排除非家族性病因 从青少年心肌病伴传导系统异常看,非家族性、有潜在病因的心肌病包括心肌炎、快速心律失常性心肌病、高嗜酸性粒细胞心肌病、结缔组织疾病累及心肌、内分泌疾病导致的心肌病(甲功异常、内分泌性高血压、肢端肥大等)、营养障碍导致的心肌病、心肌毒性药物使用和纵隔放疗等。本例患者病史及检查未提示上述病因。
排除结缔组织病 查体见四肢消瘦、关节突出、雷诺现象应重点排除结缔组织病如系统性硬皮病、混合性结缔组织病及系统性红斑狼疮等,但相关免疫指标均阴性,且此类疾病不能解释患者生长发育、骨骼、内分泌系统的异常表现,可排除。
锁定基因突变 基因突变导致单纯心肌受累性疾病如扩张型心肌病、致心律失常性右室心肌病也不能解释患者的多系统症状。一些遗传代谢性疾病可能累及心肌并伴其他系统表现,包括氨基酸代谢异常、脂肪酸氧化异常、线粒体代谢异常、神经肌肉性疾病、糖原贮积病、溶酶体贮积病等,本例患者未见相关证据且上述疾病表型与本例心血管系统外表现不符。
换个角度思考,从心血管系统外表现分析,患者身材、面貌、皮下脂肪、四肢骨骼的表型特征与一种罕见疾病——早老性疾病的临床表现类似。
早老性疾病是一类基因突变相关的过早老化性疾病,其典型代表为儿童早老症(HGPS),文献报告的发病率为1/400万~800万,致病基因是编码核纤层蛋白laminA/C的LMNA,突变位点在第11外显子c.1824C>T(p.G608G)。迄今报告的LMNA所致突变已超过100种,表型各异,统称为核纤层蛋白病(laminopathies)。核纤层蛋白病根据表型特征可分为四类——心肌病变伴或不伴骨骼肌病变、脂肪萎缩/营养不良、周围神经病变、早老症相关多系统受累。其中心肌病变多表现为左心、右心或全心扩大的扩张型心肌病,左室致密化不全或致心律失常性右室心肌病也偶有报告。约1/3心肌病变合并传导系统异常如窦房结功能异常、心房静止、房室传导阻滞等,并可进展为恶性室性心律失常、心脏骤停。本例患者的心脏表现符合核纤层蛋白病的心肌改变,而心脏外表现与儿童早老症(HGPS)的多种系统症状相吻合,如生长迟缓、毛发稀疏、皮下脂肪萎缩(以四肢为著),肢端皮肤呈硬皮样改变甚至有雷诺现象、皮肤色素沉着、小下颌、齿列不齐、梨状胸、第二性征发育不良等。但是HGPS患者病程多从婴儿期开始,平均寿命仅13岁,心血管病变特征为多发动脉粥样硬化(AS),伴高血压、高血脂及糖代谢异常,心梗和卒中是常见死亡原因。本例患者病情发生略晚,进展较慢,不伴AS及血压、血脂、血糖异常。这种表型符合同样由LMNA基因突变导致的另一种早老性疾病——不典型早老症,其临床多系统受累表现与HGPS类似,但发病较迟(幼儿期发病),寿命可达20岁或更长,心血管系统异常以心肌病变、瓣膜病多见。
治疗
根据患者表型特征、对患者及父母LMNA基因12个外显子进行测序及比对。LMNA第1外显子发现杂合突变c.175C>CG(p.Leu59Val),其父母未发现类似突变。McPhersonE等曾报告2例高加索女性病例,其临床表现与本例类似,基因突变位点略有差异(c.176T>G),但均导致第59位氨基酸改变,故本例携带突变基因的致病性得以确认。
目前尚无针对病因的有效治疗手段。文献报告LMNA基因突变导致的扩张型心肌病伴或不伴传导系统异常者恶性室性心律失常、心源性猝死发生率>50%,建议预防性置入埋藏式心律转复除颤器(ICD)。根据患者经济状况,置入心室按需型(VVI)起搏器、给予抗心衰药物治疗。妇产科建议补充激素替代治疗,患者拒绝。
# 结语
核纤层蛋白病是一类因LMNA基因突变导致的异质性疾病,可表现为心肌病变、骨骼肌营养不良、脂肪萎缩/营养不良、外周神经疾病及早老性疾病。LMNA基因突变累及心脏以扩张型心肌病为主,易合并传导系统异常,恶性室性心律失常或心脏骤停的发生率较高。在早老性疾病谱中,心血管系统病变还包括AS导致的心脑血管意外。早老性疾病是核纤层蛋白病中累及系统最为广泛的一类,会出现生长发育、内分泌代谢、骨骼、皮肤黏膜、脂肪等多系统异常。在临床工作中,对年轻的扩心病尤其是合并传导系统异常的患者,应警惕核纤层蛋白病的可能,并应注意是否存在其他组织器官受累表现,进行LMNA基因检测可以帮助确诊。
追随心脏外线索是破解谜题的关键
北京协和医院 方理刚
心肌病病因诊断很重要,直接影响患者治疗和预后。一部分心肌病和心力衰竭是可逆的,如酒精性心肌病患者,戒酒后心脏结构和功能可以恢复正常。应激性心肌病经过支持治疗后大都在短期内恢复,而有的心肌病预后很差,如心肌淀粉样变。然而,明确病因却常常很困难,继发性心肌病的诊断常通过心血管系统外的其他临床表现来得到线索。
本例为罕见病例,年轻女性,以全心衰、右心增大、缓慢心律为心血管受累的突出表现。病因分析未发现甲状腺、肾上腺疾病及自身免疫病等常见疾病证据,亦无遗传代谢病证据,似乎诊断陷入困难。但从一元论考虑,除心脏异常表现外,患者同时存在营养不良、生长发育迟缓、第二性征发育不良、面容异常、四肢骨骼异常等,促使临床医生考虑到早老性疾病可能性。早老性疾病可以解释患者心脏异常表现,进一步基因检测示LMNA基因突变,支持本病诊断。
大量研究表明,不同的心肌病表型可由同一种基因突变引起,而某一种心肌病表型可由不同基因突变引起(如家族性肥厚型心肌病),对影像学检查不能明确病因或有心肌病家族史的患者,基因检测常能提供重要诊断依据。本例患者编码核纤层蛋白laminA/C的LMNA基因突变导致不典型早老症,是引起心肌病、心衰及传导异常的根本原因,而三尖瓣重度反流加重右心扩大,持续缓慢心律亦可能是心脏扩大的因素之一。
对于早老性疾病尚缺乏针对病因的治疗。因本患者存在心脏猝死的高风险,器械治疗在本例患者有适应证。最终因经济原因,患者只置入了VVI起搏器,一定程度上可减少心脏停搏风险,并使β受体阻滞剂得以使用。积极改善患者的心室功能是重要治疗,因此只要没有禁忌证,应使用β-受体阻滞剂、血管紧张素转换酶抑制剂及醛固酮受体拮抗剂,但对于缓慢心律的心衰患者,β-受体阻滞剂的收益如何缺乏确切证据。因此本患者药物使用剂量的滴定、疗效尚需观察。对患者进行规律随访很重要,以保证规范化抗心衰治疗、了解起搏器工作状况、对患者进行心理疏导,尽可能延长患者生存期和改善生活质量。
面对疑问,不轻易止步
北京协和医院 严晓伟
患者为年轻女性,因心脏扩大、全心衰、结性心律入住我院心内科病房。单纯从心脏症状看是扩张型心肌病的临床表现。对大多数心内科医生来说,可能会止步于原发性扩张型心肌病的诊断,常规给予强心、利尿治疗,待病情平稳后加之肾素-血管紧张素系统(RAS)阻滞剂、β-受体阻滞剂和醛固酮拮抗剂治疗后让患者出院门诊随访。但本院的年轻主治医生并没有止步,没有停止思索。
患者自12岁出现雷诺现象,近半年明显脱发。首先考虑是否为风湿免疫性疾病累及心脏。我院对患者进行全套自身抗体和血管炎检测指标检查后结果均为阴性,血清补体、ESR和CRP亦均在正常范围。再回顾患者临床特点——第二性征发育不良、卵巢早衰、严重消瘦,显然风湿免疫性疾病难以解释“罪魁祸首”的全貌。
患者体重30 kg,BMI低至12.49 kg/m2;皮肤干燥,头发稀疏无光泽,双下肢水肿。心肌病变是否由严重营养不良所致?答案是否定的。因为患者血红蛋白、白蛋白水平均在正常范围,故很容易排除营养不良性心肌病的诊断。
至于高嗜酸粒细胞心肌病、内分泌疾病导致的心肌病、药物性心肌病、放射性心肌病、氨基酸代谢异常、脂肪酸氧化异常、线粒体代谢异常、神经肌肉性疾病、糖原贮积病、溶酶体贮积病等引起的心肌病变,结合患者的病史和整体临床表现,均不难排除。
通过对文献的进一步检索,罕见的核纤层蛋白病浮出水面。正如病例分析中介绍的那样,本例患者的心脏和心外表现与核纤层蛋白病有很多相似之处。但如果诊断仅止步于临床表现的相似并无任何实际意义,因为该病需要基因检测确诊。最终,在本院临床遗传实验室老师的帮助下,明确了LMNA第1外显子的杂合突变c.175C>CG。至此,这一罕见疾病累及心脏的完整病例在眼前一一呈现。
尽管对患者来说,疾病确诊并没有带来有效的治疗手段,也无法改变这位年轻患者的命运。但对临床医生来说,是向着真理又迈进了一步。更重要的是,在疾病诊治中抽丝剥茧的临床思维的培养、不轻言放弃的精神、以及在同情心驱使下的不断求索,是协和老一辈医学大师的言传身教。今天,协和的年轻一代正在用自己的行动将它们发扬光大。
精选30个心内经典病例,由专家解析并点评。12月1日起,每日发布一例,并提供PDF全文下载。反复研读,受益匪浅!稀缺专题,值得收藏!




中国医学论坛报版权所有,转载须授权
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017