


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




邢铭辉1,2 许卉妍2 综述 李文斌2 审校
DOI:10.3969/j.issn.1006-298X.2024.03.011
[基金项目]山东省自然科学基金项目(ZR202211240343);中国初级卫生保健基金会终末期肾病科研基金项目 (HT202112220001)
[作者单位]1山东第二医科大学(潍坊,261053);2山东第一医科大学第一附属医院(山东省千佛山医院)肾病学科
摘 要
肾脏纤维化是慢性肾脏病(CKD)进展至终末期肾病的共同病理特征,明确肾脏纤维化发生的分子机制对CKD的防治至关重要。内皮细胞表层的多糖蛋白复合物——糖萼组成了血液和血管壁之间最基本的屏障。最新研究显示,糖萼在肾脏纤维化过程中发挥重要作用。本文就糖萼参与肾脏纤维化发生的相关研究进展作一综述,为其防治提供新的治疗策略。
关键词 慢性肾脏病 肾脏纤维化 糖萼
Mechanism of glycocalyx involved in renal interstitial fibrosis
XING Minghui1,2,XU Huiyan2,LI Wenbin2
1Shandong Second Medical University, Weifang 261053, China
2Department of Nephrology, Shandong Provincial Qianfoshan Hospital, The First Affiliated Hospital of Shandong First Medical University, Jinan 250014, China
ABSTRACT
Renal fibrosis is a common outcome of various causes leading to the development of chronic kidney disease (CKD) to end-stage renal disease. It's crucial for the treatment of CKD patients to clarify the molecular mechanisms of renal fibrosis. There is a layer of polysaccharide protein complex (also called glycocalyx) on the endothelial cell surface, which constitutes the basic barrier between the blood and the vessel wall. Recent studies have shown that the glycocalyx degradation plays an important role in promoting renal fibrosis. This review will discuss the research progress on the mechanisms of glycocalyx involved in renal interstitial fibrosis and may provide new therapeutic strategies for CKD.
Key words chronic kidney disease renal fibrosis glycocalyx
肾脏纤维化病理过程复杂,主要表现为肾小球硬化、肾小管间质纤维化、炎症细胞浸润、微血管减少以及固有细胞丢失等多种机制[1],糖萼是由血管内皮细胞合成且覆盖其表面的多糖蛋白复合物,对内皮稳态及功能的调节发挥重要作用[2],糖萼结构和功能的异常导致肾小球毛细血管通透性增加,糖萼进而刺激肾小管上皮细胞,发生肾脏纤维化。本中心前期研究发现尿毒症患者血清中糖萼的脱落产物透明质酸和多配体蛋白聚糖(syndecan)-1表达升高[3],本文将从糖萼的组成、功能、降解的机制以及糖萼在肾脏纤维化中的研究进展进行综述。
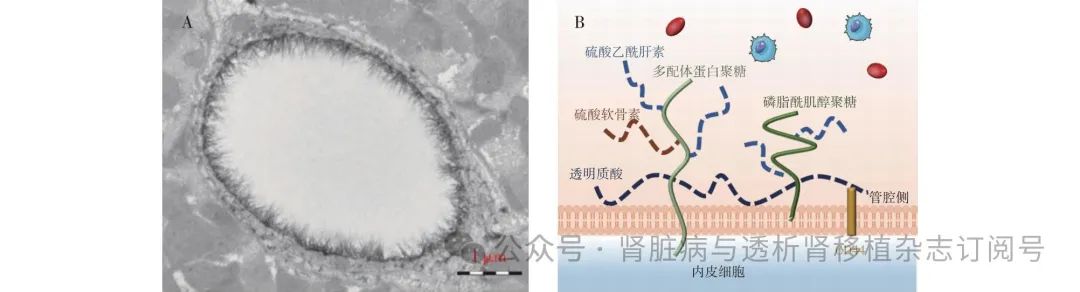
糖萼的结构 糖萼是由蛋白聚糖(proteoglycans, PG)、糖胺聚糖(glycosaminoglycan, GAG)、膜糖蛋白及吸附的血浆蛋白组成的筛网状结构。PG是由一条或多条糖胺聚糖链连接的核心蛋白质构成,主要包括syndecan1-4、磷脂酰肌醇聚糖(glypicans)和唾液酸等。GAG是一种双糖线性聚合物,有五种类型,包括硫酸乙酰肝素(heparan sulfate, HS)、硫酸软骨素(chondroitin sulfate, CS)、硫酸皮肤素(dermatan sulfate, DS)、硫酸角质素(keratan sulfate, KS)和透明质酸(hyaluronan, HA)[4]。其中占比最大的是HS,为50%~90%[5]。PG及GAG与膜链接的糖蛋白构成致密的内基质层,形成对血浆大分子的主要选择性屏障,外层主要由GAG与血浆蛋白构成,其延伸到血管腔中一个或多个微米,支持血细胞持续运动[5] (图1)。
图1 糖萼病理图片(A)[6]及糖萼成分示意图(B)
A:电子显微镜观察阿利新蓝8GX染色山羊冠状动脉毛细血管糖萼;B:生理条件下,内皮细胞表面覆盖多功能聚糖——糖萼,由蛋白聚糖 (多配体蛋白聚糖和磷脂酰肌醇聚糖)、糖胺聚糖(硫酸乙酰肝素、透明质酸、硫酸软骨素、硫酸皮肤素、硫酸角质素)以及吸附的血浆蛋白构成
糖萼的功能 糖萼的结构与内皮细胞生理功能息息相关,包括血管通透性、凝血及炎症反应等。糖萼位于血流和内皮细胞之间,是血管通透性的重要决定因素,GAG的高度硫酸化,使其呈现出负电荷的特性,形成白蛋白等血浆蛋白的电荷屏障。糖萼还影响血细胞与血管壁的相互作用,它含有多种内皮细胞黏附分子,同时减弱了白细胞对这些分子的黏附。血管内皮细胞常常暴露于血流的机械力,剪切应力的暴露导致内皮细胞产生一氧化氮(NO),这是血管张力的重要决定因素,糖萼可作为内皮细胞剪切应力的机械传感器,研究证明HS和HA是将生物力学转化为生物化学信号的重要分子[5]。
多种刺激因子可导致糖萼的结构和功能紊乱。通过测量糖萼的厚度或者检测循环中糖萼的成分(Syndecans、HS、HA等)来判断糖萼损伤,也可通过对舌下微循环进行正交偏振光谱(OPS)成像计算糖萼的体积[7]。
糖萼与疾病 在糖尿病、动脉粥样硬化、脓毒症、缺血/再灌注损伤(IRI)、慢性或急性肾脏病等[8]多种疾病中均发现糖萼结构的破坏。通过对舌下微循环进行OPS成像发现与健康对照组相比,1型糖尿病患者糖萼体积减小同时HA及其降解酶透明质酸酶水平增加[7]。高血糖是一种有效的促氧化和促炎症因子[5],通过输注强抗氧化剂N-乙酰-L-半胱氨酸可减少高血糖期间糖萼的损伤[2]。观察几何结构复杂的血管发现,暴露于血流速度不均匀的血管内皮细胞糖萼层变薄[9]。脓毒症时,活性氧与促炎因子[如肿瘤坏死因子α(TNF-α)、白细胞介素(IL)1β]能够激活基质金属蛋白酶、乙酰肝素酶和透明质酸酶等介导的炎症机制[10],导致糖萼降解增加。Ilyina等[11]证实脓毒性休克患者体内糖萼的脱落产物syndecan-1水平升高。
在慢性肾脏病(CKD)患者血清中糖萼脱落物(HA、syndecan-1)随疾病的进展而增高[12]。同时,研究发现活动期狼疮性肾炎患者血清中syndecan-1的表达水平增加,其表达与抗ds-DNA抗体滴度、蛋白尿、血清肌酐水平及系统性红斑狼疮疾病活动度评分相关[13]。糖萼作为肾小球滤过屏障的重要组成成分,其结构及功能的破坏被认为是导致蛋白尿发生的重要机制,Nieuwdorp等[7]发现1型糖尿病和蛋白尿患者的内皮糖萼体积减小,减小的程度与尿白蛋白水平直接相关。
糖萼结构破坏使循环中的白细胞更容易黏附在血管内皮细胞上,促进炎症的发展,也使内皮通透性增加,机械传导受损,抗凝剂减少等,致使内皮细胞功能障碍[8],促进肾脏纤维化的发生发展。
生理条件下,内皮糖萼层与流动的血液成分处于动态平衡。在肾脏病患者中,慢性炎症状态、TNF-α、内毒素及氧化低密度脂蛋白等多种因素可导致糖萼损伤,进而导致内皮细胞功能障碍,使糖萼成分进入肾小管管腔中并作用于肾小管上皮细胞,促进肾小管间质纤维化,参与肾脏纤维化[2, 14]。其中,HS、HA及Syndecan-4作为糖萼的重要成分在肾纤维化进展中起重要作用。
硫酸乙酰肝素(HS)
HS硫酸化 HS能够促进、调节白细胞募集,与转化生长因子β(TGF-β)等促纤维化因子结合,在肾脏纤维化的发展中至关重要[15]。
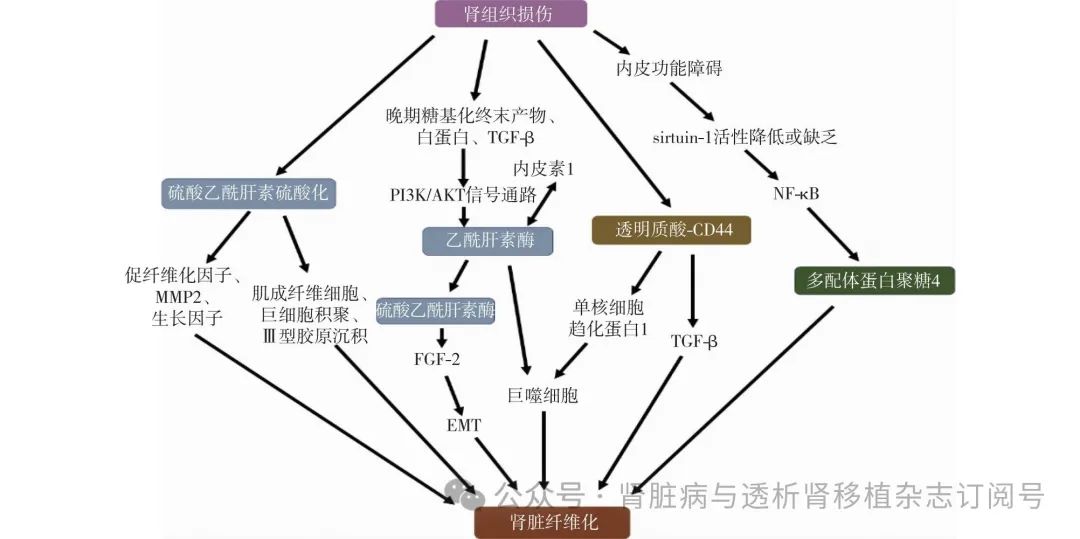
图2 糖萼参与肾脏纤维化机制图
TGF-β:转化生长因子β;MMP2:基质金属蛋白酶2;PI3K/AKT:磷脂酰肌醇3-激酶/蛋白激酶B;FGF-2:成纤维细胞生长因子2;EMT:上皮-间质转化;NF-κB:核因子κB;硫酸乙酰肝素硫酸化诱导炎症细胞浸润、促进肌成纤维细胞积聚以及促纤维化因子等的释放。一系列促纤维化因素通过激活PI3K/AKT信号通路上调乙酰肝素酶的表达,乙酰肝素酶过表达触发硫酸乙酰肝素降解并调节syndecan-1表达;syndecan-1的可溶性硫酸乙酰肝素片段是FGF-2有效的信号激活剂,从而促进管状细胞EMT。乙酰肝素酶的产生还通过与内皮素-1的恶性循环来维持。透明质酸透过诱导巨噬细胞募集以及增强TGF-β通路发生肾脏纤维化。sirtuin-1的缺乏通过NF-κB 炎症通路使多配体蛋白聚糖-4表达释放增加进而诱导纤维化
HS可被磺基转移酶修饰硫酸化。在实验中使用将硫酸化修饰相关基因N-脱乙酰基酶/N-磺基转移酶1(Ndst1)特异性消除的小鼠诱导糖尿病模型,与对照组相比,内皮Ndst1缺乏的小鼠HS硫酸化受抑制,Ⅲ型胶原在肾小管间质和肾小球中的沉积,肌成纤维细胞在肾小管-间质中的积聚减少;同时,抑制HS硫酸化后,肾小球及肾小管间质中巨噬细胞积聚、CXCL16介导的纤维细胞迁移改善。故而HS硫酸化在纤维化发展中起重要作用[16]。发生慢性同种异体移植物排斥反应的肾移植患者及单侧输尿管梗阻(UUO)模型中均发现肾小管上皮细胞中HS6-O-硫酸化结构域表达增加,HS6-O-硫酸化作为共受体使生长因子与其同源酪氨酸激酶成纤维细胞生长因子2(FGF-2)受体结合调节FGF2信号传导,导致促纤维化细胞因子(例如TGF-β)、MMP2和生长因子的进一步产生[17]。以上研究结果表明HS硫酸化通过诱导炎症细胞浸润、促进肌成纤维细胞积聚等机制参与肾脏纤维化,抑制HS硫酸化是可能的肾纤维化的干预途径。
乙酰肝素酶(HPSE) 糖萼硫酸乙酰肝素修饰酶的表达与纤维化的发展显著相关,HPSE表现出最强的相关性。HPSE具有调节多种机制的能力,包括肾小球选择通透性、肾小管上皮-间质转化(EMT)以及炎症浸润等,使其在纤维发展中发挥具有重要作用,因此其作为治疗靶点已得到广泛认可。HPSE不仅可以修饰HS,也可直接影响FGF-2、TGF-β、内皮素1(ET-1)等分子发挥生物效应。Masola等[18]发现Roneparstat可通过抑制HPSE减轻IRI的程度,阻止纤维化的发展。
降解HS 有研究证实,多种肾损伤动物模型及肾脏病患者的肾组织中HPSE过度表达[19-20]。由于HPSE的表达与肾小球基膜HS的含量呈负相关,因此,Van Den Hoven等[21]认为在糖尿病肾病中HPSE对肾小球HS的降解参与蛋白尿的发生。一些促纤维化因素如蛋白尿、高葡萄糖浓度、晚期糖基化终产物、TGF-β和FGF-2等可通过激活磷脂酰肌醇3-激酶/蛋白激酶B(PI3K/AKT)上调HPSE表达,PI3K/AKT是一种参与肾小管细胞EMT的信号通路[19]。HPSE的过表达可以触发HS降解并调节syndecan-1的表达,syndecan-1的可溶性HS片段是FGF-2 信号通路强有力激活剂,从而促进肾小管细胞EMT过程[22]。
上调FGF-2和TGF-β的作用 Masola等[19]研究表明,当肾小管细胞缺乏HPSE时,FGF-2和TGF-β不能介导其产生EMT。这主要是由于Syndecan-1在HPSE沉默的细胞表面上的过度表达,细胞与FGF-2的亲和力较低;在缺乏HPSE的肾小管细胞中,PI3K/AKT通路的继发性损伤也危及FGF-2诱导的EMT。体外实验证实,缺乏HPSE可以阻止受损的肾小管细胞TGF-β过表达;也有研究证实糖尿病HPSE-Ko小鼠的肾组织中没有TGF-β的增加,也并未出现纤维化。因此,HPSE对FGF-2和TGF-β诱导管状细胞的EMT是必要的。
调节炎症反应 Gil等[23]证明,与野生型动物不同,HPSE敲除小鼠在诱导1型糖尿病后,肾实质中无明显的F4/80阳性巨噬细胞浸润。研究证实,HPSE可在IRI的肾脏中将巨噬细胞极化为促进炎症发生和促纤维化的M1型巨噬细胞,同时上调肾小管上皮细胞和巨噬细胞Toll样受体(TLR)基因表达,增加了它们对TLR配体的敏感性,发挥促炎作用[19, 24]。肾脏是具有缺氧保护系统的器官,缺氧诱导因子(HIF)是该系统的“主基因”。在IRI期间,HIF-1α在肾小管细胞缺氧阶段上调,可能是通过PI3K/AKT途径促进EMT发生[25],抑制HPSE可阻止HIF-1α的积累间接抑制EMT的发生[26]。
增加ET-1 ET-1是一种血管收缩剂,可在内皮激活时释放,导致血管收缩,肾小球囊内压力升高,从而导致肾脏缺血,发生肾脏纤维化[18]。研究表明ET-1与HPSE之间存在相互作用,HPSE的过表达可增加IRI中ET-1的水平,ET-1也可诱导足细胞上调HPSE的表达,继而引发蛋白尿及肾功能损害[18, 27]。使用HPSE抑制剂可下调ET-1的表达并打破其恶性循环过程[18]。
目前研究虽然发现糖萼HS及其修饰酶参与肾脏纤维化的发展,但具体的分子机制仍然是不明了的,因此进一步明确其发生的分子机制对于靶点药物的开发是有必要的。
透明质酸(HA) 目前研究表明, HA分子量的不同使其在肾脏纤维化发展中呈双向性[28],高分子量透明质酸(HMW-HA)表现为抗炎及抗纤维化特性。Wang等[29]发现UUO小鼠模型中,主要是通过HA合成酶(Has1、Has2)促进HMW-HA的合成增加HA沉积,为进一步明确HMW-HA与肾纤维化的关系,该团队使用慢-IL-10-GFP构建体上调IL-10的表达,促进HMW-HA的合成,发现经IL-10处理的UUO小鼠肾小管间质纤维化及α平滑肌肌动蛋白(α-SMA)表达较对照组减少。有研究证实HMW-HA的产生是对肾损伤的初始生理反应,伴随时间延长,很大一部分被降解成更小的碎片,主要以低分子量透明质酸(LMW-HA)的形式沉积,表现为促炎及促纤维化作用[29]。Colombaro等[30]研究发现,敲除小鼠透明质酸酶后,UUO小鼠肾皮质中HA较野生型小鼠积聚增多且持续时间延长,通过诱导肾小管上皮细胞MCP-1上调使巨噬细胞募集延长,进而发生肾脏纤维化,但该实验并未对HA分子量做出相关研究。研究证实,HA通过与CD44相互作用诱导肾小管细胞刺激单核细胞趋化蛋白1(MCP-1),HA-CD44相互作用增强了TGF-β通路,导致IRI后肾脏纤维化[31],阻断HA和CD44之间的相互作用可能是阻止纤维化进展的重要途径。目前HA在纤维化发展中作用机制的研究仍然较少,对HA的干预可能是抗纤维化的重要方向之一。
多配体蛋白聚糖4(syndecan-4) 目前证实syndecan-4胞外结构是在内皮细胞障碍基础上,作为激活成纤维细胞的信号分子以及诱导巨噬细胞募集,引起细胞外基质堆积进而发生肾纤维化[32]。
已知内皮功能障碍是CKD患者的中心事件之一,在 CKD 患者中sirtuin-1可能活性降低甚至缺乏,内皮细胞sirtuin-1缺陷型(Sirt1endo-/-)小鼠是研究内皮功能障碍的常用模型。Lipphardt等[32]用 TGF-β 诱导Sirt1endo-/-小鼠的肾微血管内皮细胞(RMVEC)发现syndecan-4表达增加,syndecan-4水平的升高归因于间质中其胞外结构的积累。为了进一步阐明syndecan-4胞外域与肾纤维化的关系,该团队在小鼠左肾包膜下注射syndecan-4胞外结构域,发现sirtuin-1缺乏小鼠是通过激活核因子κB 依赖性炎症通路,使P65蛋白去乙酰化减少,导致其释放和核易位从而诱导 Syndecan-4 的表达,并使得氧化应激增加,进而激活去整合素基质金属蛋白酶17(ADAM-17)等,导致syndecan-4 脱落增强,间质纤维化较前增加。以上结果表明sirtuin-1在内皮糖萼保护中起到核心作用,目前已有3种小分子sirtuin-1激活剂(SRT2104、SRT2379和SRT3025)在临床试验中进行了测试[2]。研究证实,在老年志愿者、健康吸烟者和2型糖尿病患者中,化合物SRT2104对脂质、降低血清胆固醇、低密度脂蛋白水平和三酰甘油有益。不仅如此,SRT2104还减少了脂多糖(LPS)诱导的炎症介质释放,因此这3种化合物作为抗纤维化药物仍需有利的证据及深入机制的探讨(图2)[33]。
小结:CKD患者中存在多种机制导致糖萼受损,而糖萼成分可能通过与促纤维化因子相互作用、炎症、氧化应激等参与肾纤维化发生,进一步探索保护糖萼完整性将作为干预靶点来防治肾脏纤维化的发生发展,为延缓CKD患者肾功能恶化提供新的治疗策略。
参考文献


来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017