



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




慢性肾脏病(CKD)已经成为全球性公共健康问题,其患病率和死亡率较高。CKD可由多种疾病过程引起,其病因往往难以用传统的临床诊断方法确定。大多数CKD病例是由糖尿病或高血压引起的,近年来,CKD的遗传因素也逐渐受到重视。改善全球肾脏病预后组织(KDIGO)强调了遗传学在CKD的分类和管理中的重要性,并建议临床医生考虑对CKD患者进行基因检测,以提高诊断的准确性。
2024年8月14日来自Edmond and Lily Safra儿童医院的Asaf Vivante团队在《新英格兰医学杂志》上发表题为“Genetics of Chronic Kidney Disease”的综述,讨论了CKD的遗传学诊断和治疗方法,重点分析了单基因遗传CKD和具有进展性CKD风险的变异。
CKD的流行病学
目前已知许多常见的CKD病因有家族聚集的特点,不同种族和民族群体之间的差异也体现了遗传因素的影响。多份报告表明,多种不同的单基因疾病可以解释大约30%-50%的儿童CKD病例和约10%-20%的成人病例。
CKD的分类
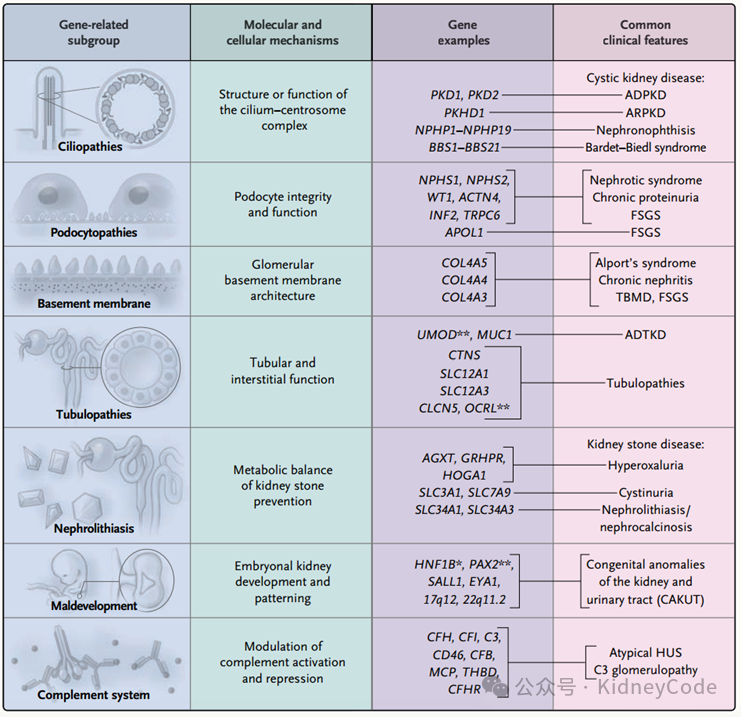
CKD的遗传风险可在低外显率到高外显率的遗传变异谱上观察到,CKD的病因可根据临床表现、肾脏组织学特征或遗传特征进行分类,而CKD的遗传和表型异质性使基因相关的定义更为准确。遗传诊断指出了特定疾病的根本原因,提供了对分子机制的见解,且可能导致10%-50%病例的临床或组织学诊断被重新分类。因此,基因检测可以补充甚至取代肾活检,成为诊断和治疗某些形式CKD的标准,且CKD的分子分类方式可能促进精准医学在临床实践中的发展。(图1)
图1 CKD遗传来源的主要细胞和分子类型
遗传性CKD的基因型及表型
1.多囊肾病
多囊肾病是一种由纤毛-中心体复合物内蛋白质改变引起的多种遗传疾病。其中,ADPKD是最常见的遗传性肾病,由PKD1或PKD2基因突变引起。目前已知超过15种不同基因突变可以模拟ADPKD表型,目前使用PKD1的Sanger测序或短读外显子组测序进行基因检测具有一定的局限性,它们与相似的假基因存在重复和同源性,因此还需通过PCR来验证PKD1的变异。肾消耗病是异质性常染色体隐性遗传病,其最准确的诊断方法是基因检测,诊断率约为70%,其中NPHP1的纯合变异占20%-50%。其他病例是由大约90种不同基因中的任何一种突变引起,这些基因与调节细胞极性、DNA损伤反应或循环AMP信号的多种分子途径有关。
2.遗传性肾小球疾病
遗传性肾小球疾病通常由维持足细胞功能的基因突变或构成肾小球基底膜蛋白质的致病性变异引起。其中Alport综合征是由于编码IV型胶原蛋白α3、α4和α5链发生基因突变,错义突变会影响组装胶原异三聚体结构所需的甘氨酸残基,因此建立IV型胶原编码基因突变的精确分子诊断对于疾病的治疗至关重要。另外,在先天性常染色体隐性肾病综合征患者中发现了NPHS1和NPHS2等足细胞相关基因突变,已知超过50种不同基因产物可以使足细胞超微结构发生改变,介导信号转导或控制足细胞细胞骨架的重排。
3.肾小管遗传病
影响肾小管功能导致的遗传性疾病包括50多种综合征,通常涉及离子通道或转运体的异常。尿调节素(UMOD)是一种肾特异性蛋白,由UMOD变异引起一系列疾病称为UMOD相关常染色体显性小管间质肾病(ADTKD)。大多数致病突变是错义突变,60%含有半胱氨酸残留致病性UMOD变异导致蛋白质错误折叠,随后蛋白质滞留在内质网中,并在亨利环的厚升肢中错误靶向尿调蛋白。由此引起小管间质损伤最终会导致CKD。
4.肾结石
肾结石病属于多因素疾病,由于代谢不平衡导致尿结晶引起。编码维持代谢平衡产物的基因突变引起遗传性肾结石,如腺嘌呤磷酸核糖基转移酶缺乏症、家族性低镁血症伴高钙尿症等常导致CKD并进展为肾衰竭。
5.具有复杂遗传基础的肾病
还有一些具有复杂遗传基础的肾病,例APOL1相关CKD。APOL1的基因型-表型相关性十分独特,在常染色体隐性遗传病中经典孟德尔高外显率在APOL1相关CKD中不存在。此外,精液全基因组关联研究确定了活检证实的特发性膜性肾病和IgA肾病的遗传基础,发现每一种免疫介导的肾小球病变都有一个疾病特异性风险位点,可能揭示其病理生理机制。
6.原因不明的慢性肾病
在美国和欧洲的成人肾衰竭病例中,大约有10%-20%的病例在经过大量检查后仍不清楚CKD的病因,这些病例中高达20%可归因于遗传条件。
CKD患者遗传学诊断及临床意义
建立遗传诊断首先需要综合表型,获得详细的家族史及三代谱系。CKD基因检测的诊断率取决于疾病表型、种族、血缘、家族史和发病年龄等。依照一系列相关适应证,医生可对CKD患者进行遗传分析。肾脏疾病的单基因分析主要通过大型基因面板、外显子组测序和全基因组测序等方法。CKD患者通过基因诊断能够确定明确病因,避免了肾脏活检的需要,同时可以为不同类型的患者提供更有针对性的治疗方案。
CKD新兴治疗方法
基因诊断确定了有效的基因治疗资格,使CKD患者成为新兴治疗的候选人。例如使用小分子inaxaplin治疗APOL1相关CKD,逆转了APOL1高危变异的毒性获得,阻止了APOL1相关CKD的进展。此外,确定CKD单基因条件有助于制定有针对性的分子治疗策略。例如在PKD1缺失的小鼠中,通过PKD1转基因转移重新表达PC1蛋白可显著延缓囊肿的发育。另一种方法是使用基于RNA技术,包括化学修饰的反义寡核苷酸或小干扰RNA,它们可以调节细胞RNA的丰度、加工和翻译,不需要将转基因传递到肾脏。目前也正在研究其他基于基因的CKD治疗方法,但药物递送到特定的肾细胞仍然具有挑战性。
文章结论与展望
综上,这篇综述讨论了遗传性CKD的诊断和治疗方法。发现基因检测在CKD临床治疗试验中的应用越来越广泛,并且可能通过基因特异性治疗改变肾脏病学的发展,而遗传因素在肾脏供者中的作用还需要进一步研究。研究团队还应继续扩大对代表性不足的患病人群的遗传信息采集,并不断开发新的基因治疗手段,以减轻CKD在全球范围内造成的巨大负担。
来源:KidneyCode
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017