


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




胰腺癌是一种侵袭力强、致死率高的恶性肿瘤,由于其发病隐匿,进展迅速,预后不良,胰腺癌在癌症相关死亡原因中位居前列。2021年中国国家癌症中心统计数据显示,胰腺癌位居我国男性恶性肿瘤发病率第7位,女性第11位,占恶性肿瘤相关死亡率第6位。2021年美国统计数据显示,在所有恶性肿瘤中,胰腺癌新发病例男性居第10位,女性居第8位;死亡率在男性、女性中均位居第4位。2020年,美国有57 600例新发胰腺癌病例,其中47 050例死亡[1]。胰腺导管腺癌(pancreatic ductal adenocarcinoma, PDAC)是最常见的胰腺癌类型,2018年数据显示,PDAC患者的5年生存率仅为9%[2]。目前,手术切除仍是治疗胰腺癌的唯一方法,但只有20%患者符合指征而接受外科手术,即使手术成功切除者,超过80%患者最终仍会发生局部复发或转移[3]。近期,一线化疗手段在提高PDAC患者生存率方面取得重要突破,然而二线或后期治疗中仍然缺乏高效的治疗方案。此外,作为治疗癌症的新兴策略,使用检查点抑制剂的单一疗法对PDAC几乎完全无效[4],这主要是由PDAC免疫抑制肿瘤微环境所致。综上所述,针对PDAC的治疗需寻求新突破。
肿瘤代谢相关的基因突变被认为是胰腺癌进展和预后不良的基础。KRAS致癌突变是胰腺癌中最常见的基因突变,超过90%PDAC患者可检测出KRAS突变[5]。KRAS突变损害蛋白内在三磷酸鸟苷(GTP)酶活性,阻止其从活性形式GTP向非活性形式二磷酸鸟苷(GDP)的转化,使KRAS蛋白始终与GTP结合而被永久激活,并作为分子开关激活下游多种细胞内信号通路和转录因子,诱导细胞增殖、迁移、转化和存活。本文将重点介绍由突变型KRAS介导的肿瘤代谢改变在胰腺癌发生和进展中的作用,以及靶向KRAS治疗策略的研究现状与前景。

不同类型肿瘤代谢差异显著,这是由特定的基因突变、组织来源或肿瘤微环境决定的。PDAC肿瘤微环境的特点是缺氧、低营养水平、高间质压力和结缔组织增生。其肿瘤微环境富含致密纤维,主要由细胞外基质(如胶原蛋白和透明质酸)组成[6]。除了透明质酸、细胞因子、趋化因子和多种胶原蛋白外,PDAC的肿瘤微环境中还包含多种细胞成分,如巨噬细胞、树突细胞、T淋巴细胞和B淋巴细胞[7]。局部免疫抑制为肿瘤发生、进展以及远处转移提供了理想环境。
以CD4+调节性T淋巴细胞为主的“冷”肿瘤通常会逃避免疫系统;致密结缔组织增生阻碍了治疗药物的局部摄取。致密间质压迫血管,使得血管形成受阻,灌注不足,导致肿瘤内严重持续缺氧[8]。但PDAC细胞却能在供氧不足、营养匮乏的条件下展现出非凡的生长优势,这依赖于其独特的代谢途径:(1)对细胞内营养物质的能量代谢进行重编程,包括葡萄糖、氨基酸和脂质;(2)通过清除和循环利用来改善养分的摄取;(3)与微环境中的其他成分交联互通,进行代谢调控。而在不利微环境下的适应性突变(或选择)则是肿瘤生存的关键。KRAS高突变发生率促使人们探讨其对胰腺癌侵袭性、转移性及代谢重编程的影响。
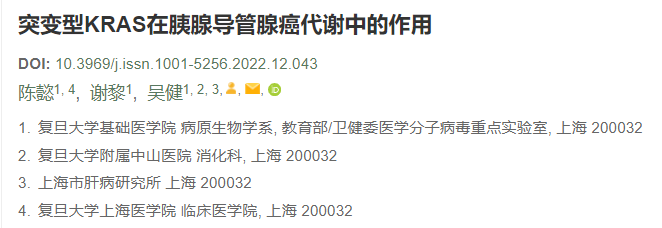
KRAS基因编码蛋白质KRAS,一种小GTP酶。它通过偶联细胞膜生长因子受体与细胞内信号通路和转录因子,充当多种细胞过程的“分子开关”。KRAS蛋白含有两个功能域:G结构域和细胞膜锚定域,G结构域参与结合和水解GTP,细胞膜锚定域C端由CAAX基序构成,这一结构对膜锚定作用很重要。KRAS与GTP结合时被激活,与GDP结合时失活。一旦KRAS蛋白与GTP结合,它会与下游80多个效应蛋白和信号通路相互作用,包括丝裂原活化蛋白激酶-MAPK激酶通路(MAPK-MEK通路)、磷脂酰肌醇3激酶-蛋白激酶B-雷帕霉素靶蛋白通路(PI3K-AKT-mTOR通路)、加速性纤维肉瘤-MAPK激酶-细胞外调节蛋白激酶通路(RAF-MEK-ERK通路)等。核转录因子如ELK、JUN和MYC也会被激活,进而刺激细胞分化、增殖、迁移、转化、黏附和存活[9-10]。
KRAS基因第12位密码子上的激活点突变是大部分胰腺癌病例(70%~95%)的起始事件,其中KRASG12D较为常见。KRAS的点突变破坏了RAS的内在GTP酶活性,使GTP酶活化蛋白(GAP)失去对GTP的失活作用(即促进GTP向GDP转化),因此KRAS蛋白与GTP永久结合,持续激活下游信号通路,维持细胞存活和增殖[10](图 1)。
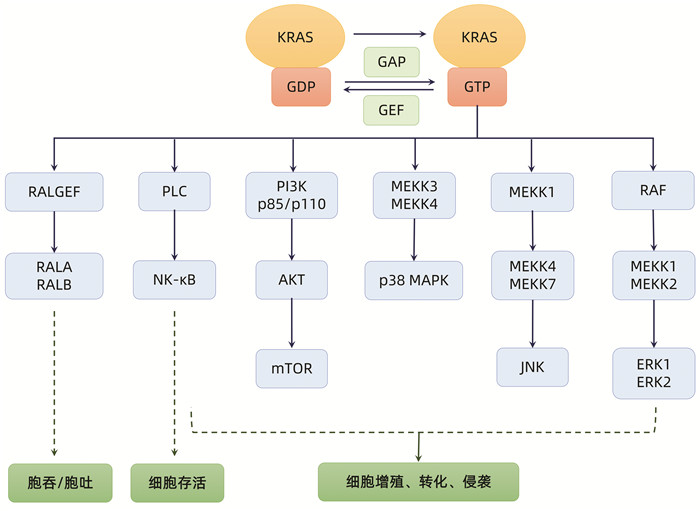
图 1 KRAS信号通路及其对细胞功能的作用
注:KRAS在与GTP结合时被激活,启动下游多种信号通路,最终促进细胞存活、增殖、转化、侵袭、胞吞/胞吐功能。KRAS的突变破坏RAS的内在GTP酶活性,使GAP失去对GTP的失活作用,促进GTP向GDP转化,导致KRAS及其下游信号通路的持续激活。GEF:鸟苷酸交换因子;RALGEF:鸟嘌呤核苷酸交换因子;RALA:Ras样蛋白A;RALB:Ras样蛋白B;PLC:磷酯酶C;NK-κB:核因子κB;MEKK1/2/3/4:MAPK激酶1/2/3/4;JNK:c-Jun氨基末端激酶。
PDAC肿瘤微环境表现为缺氧、低营养水平、高间质压力和结缔组织增生。为了在这种恶劣微环境中生存,PDAC肿瘤细胞改变代谢通路,参与葡萄糖、氨基酸、脂质代谢的重编程;利用溶酶体清除途径,如细胞自噬和巨胞饮作用(macropinocytosis),获取生存生长的“燃料”,支持、维护代谢稳态;PDAC的肿瘤微环境中多种细胞成分,如癌症相关成纤维细胞(cancer-associated fibroblasts, CAF)、神经元和免疫细胞,在营养限制的条件下支撑PDAC的生长[11]。
在PDAC中,致癌突变型KRAS与葡萄糖摄取增加、转向合成代谢途径分流、谷氨酰胺重编程以及活性氧(ROS)调控有关[12]。此外,为了解决营养稀缺或供应不平衡,致癌突变型KRAS激活独特的代谢清除途径。2020年的研究[9, 13]表明,在PDAC小鼠模型中,致癌突变型KRAS通过上调细胞因子,促进免疫细胞浸润,重新编程PDAC肿瘤代谢。
Warburg效应是肿瘤细胞代谢的共同特征,表现为葡萄糖摄取增加,线粒体氧化磷酸化转变为有氧糖酵解。这种代谢变化有利于癌细胞在恶劣环境(缺氧、营养缺乏)中存活生长。突变型KRAS通过调节细胞代谢通路参与这一过程。
突变型KRAS通过增加葡萄糖转运体、糖代谢关键酶(如Hk1、Hk2、Pfk1、Ldha)、己糖胺生物合成途径(hexosamine biosynthesis pathway, HBP)的限速酶谷氨酰胺6-磷酸果糖转移酶以及非氧化性磷酸戊糖途径(pentose phosphate pathway, PPP)中5-磷酸核丁糖异构酶和5-磷酸核丁糖-3-差向异构酶的表达,提高葡萄糖摄取和乳酸产量,从而促进糖酵解通量。其机制在于突变型KRAS可持续激活下游MAPK信号通路与转录因子Myc,在转录水平调节上述基因的表达[14]。己糖激酶1、己糖激酶2、磷酸果糖激酶1以及乳酸脱氢酶A作为糖酵解的限速酶,这些酶类的编码基因表达上调有助于增强Warburg效应和糖酵解过程。HBP作为葡萄糖代谢的分支途径,为蛋白质和脂质糖基化提供了底物,这一代谢途径的增强被认为是肿瘤进展的关键。除了调控糖酵解外,突变的KRAS刺激磷酸甘油酸激酶1向线粒体易位,导致癌细胞中丙酮酸脱氢酶激酶1(PDHK1)磷酸化,磷酸化PDHK1抑制丙酮酸脱氢酶(PDH)复合物,抑制PDAC细胞中线粒体氧化磷酸化(OXPHOS)[15] (图 2)。
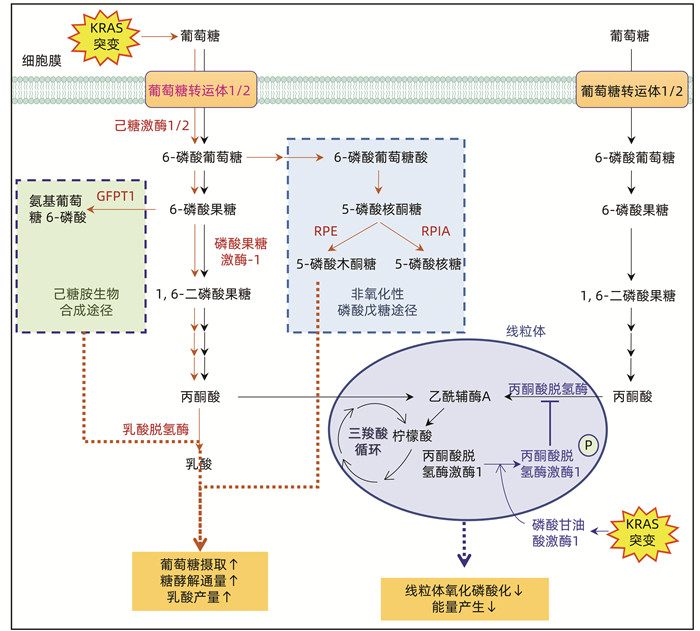
图 2 KRAS突变影响胰腺导管腺癌代谢重编程
注:葡萄糖在胞液中可裂解为丙酮酸,是葡萄糖无氧氧化和有氧氧化的共同起始途径。在正常细胞中,氧供充足,丙酮酸主要进入线粒体,经历三羧酸循环,遵循糖的有氧氧化途径(黑色箭头路线所示)。(1)KRAS突变,致使葡萄糖转运体、糖酵解限速酶、糖代谢分支途径的关键酶(棕色)上调,糖代谢以无氧氧化为主(棕色箭头路线所示),从而促进葡萄糖摄取,增加糖酵解通量。(2)KRAS突变,刺激磷酸甘油酸激酶1向线粒体易位,致使癌细胞中丙酮酸脱氢酶激酶1磷酸化,抑制丙酮酸脱氢酶(蓝色箭头路线所示),从而抑制胰腺癌细胞中线粒体氧化磷酸化,产能减少。GFPT1:谷氨酰胺6-磷酸果糖转移酶;RPE:5-磷酸核丁糖-3-差向异构酶;RPIA:5-磷酸核丁糖异构酶。
脂质代谢在癌症进展中有着至关重要的作用。与依赖膳食脂肪的正常细胞不同,肿瘤细胞中约93%的三酰甘油脂肪酸是由线粒体内柠檬酸(线粒体内乙酰辅酶A与胞质内乙酰辅酶A之间的中间体)从头合成。致癌突变型KRAS可诱导肿瘤细胞从胞外溶血磷脂摄取单不饱和脂肪酸,PDAC的缺氧肿瘤微环境对这一过程也有促进作用。此外,致癌突变型KRAS可调节激素敏感性脂肪酶,进而调控胰腺癌细胞的脂质储存[16]。
近期研究[17]显示,酰基辅酶A合成酶长链3(ACSL3)在KRAS驱动的胰腺癌中过度表达。ACSL3将细胞外不饱和脂肪酸转化为无法出胞的亲水性脂肪酰基辅酶A酯(hydrophilic fatty acyl-CoA esters),从而促进细胞外不饱和脂肪酸的活化和保存[18]。在胰腺癌的小鼠模型中,敲除基因ACSL3降低肿瘤细胞增殖,部分原因是抑制了纤溶酶原激活物抑制剂-1的生成,进而减弱了肿瘤微环境中结缔组织增生和免疫抑制[17]。瑞士伯尔尼大学的一项新近研究(2022年)[19]表明,通过限制血脂或抑制ACSL3来消耗细胞外来源的脂质会触发细胞自噬,这一过程保护PDAC肿瘤细胞避免生物能中间体的损耗。细胞外脂质供应在PDAC肿瘤细胞的脂肪酸供应中占据核心地位,发现这一肿瘤细胞代谢脆弱性对PDAC的治疗有着指导意义。
突变型KRAS对肿瘤细胞氨基酸代谢发挥调节作用。谷氨酰胺代谢作为氮和碳的主要来源,有助于大分子合成和氧化还原平衡,对癌细胞的生存至关重要。在PDAC中,致癌突变型KRAS诱导磷酸戊糖途径的代谢重组,从而使5-磷酸核糖的生物合成与NADPH的生成解耦联。这种代谢改变促使癌细胞嗜谷氨酰胺。为满足对NADPH的需求,KRAS通过转录抑制谷氨酸脱氢酶、诱导天冬氨酸转氨酶的表达来促进谷氨酰胺的代谢,维持肿瘤生长和存活。其中,线粒体各氨酰胺来源的天冬氨酸(Asp)一旦被转运到细胞质,就会转化为草酰乙酸、苹果酸,最终成为丙酮酸以生成NADPH,从而平衡细胞氧化还原稳态与大分子合成。此外,由共激活因子相关精氨酸甲基转移酶1诱导的苹果酸脱氢酶1精氨酸甲基化能抑制肿瘤生长,但KRAS的激活可以解除这一抑制,保持癌细胞的活力,促进细胞增殖[20]。
如前所述,线粒体各氨酰胺来源的Asp必须被转运到细胞质中才能进行后续的转化,因此负责转运过程的线粒体蛋白至关重要。线粒体解耦连蛋白2(UCP2)催化Asp的转运并促进肿瘤的生长。研究显示,UCP2沉默的KRASmut细胞系谷氨酰胺分解减少,NADPH/NADP+比值降低,活性氧水平升高。体内与体外实验都表明,UCP2基因沉默强烈抑制了KRAS突变型PDAC肿瘤细胞的生长[21]。UCP2以其Asp转运活性连接线粒体与细胞质反应,为致癌突变型KRAS诱导的谷氨酰胺代谢重组所必需,因此它也被视为PDAC治疗的关键代谢靶点。
巨胞饮作用是一种营养清除途径,是细胞通过内吞作用大量摄取细胞外液获得营养的过程,已被确认为营养限制条件下维持PDAC生存、生长的重要机制[22]。PDAC肿瘤细胞可通过巨胞饮作用从细胞外液中摄取细胞外蛋白(如血清白蛋白),蛋白随后被溶酶体降解,为中心碳代谢供应氨基酸。可见,巨胞饮作用对维持PDAC肿瘤细胞的氨基酸供应有着重要作用。突变型KRAS与肿瘤细胞表面的αvβ3和半乳糖凝集素3(galectin-3)形成复合物,促进癌细胞的巨胞饮作用;这一复合物也维持胰腺癌细胞的氧化还原平衡。
已有研究[23]表明,致癌性RAS诱导巨胞饮作用需要多种效应因子如PI3K信号通路、小GTP酶(Rac1、Cdc42)的活性,液泡ATP酶(V-ATPase)则是RAS依赖的巨胞饮作用的关键效应因子。研究显示,致癌突变型KRAS通过蛋白激酶A和Rac1的活性促进V-ATPase向质膜易位。
自噬是将细胞质成分运送至溶酶体进行降解的过程,它降解细胞内大分子和细胞器,循环利用细胞内生物能成分,对于维持PDAC晚期阶段的生存稳态和生长至关重要。因此,它在维持肿瘤中的能量稳态、提供代谢燃料起着关键作用。此外,自噬通过控制活性氧的产生、维持OXPHOS来促进胰腺癌的进展,升高的活性氧水平也会反过来促进胰腺癌细胞的自噬作用,这一过程受到KRAS的调节[24]。
自噬在维持PDAC免疫抑制环境中也发挥重要作用。突变型KRAS呈持续活化状态,激活yes相关蛋白(YAP)-TAZ途径及其下游Janus激酶信号转导和转录激活因子3(JAK-STAT3)信号转导,通过葡萄糖代谢重编程诱导细胞自噬相关的MHC-I降解[25-26]。研究[27]发现,PDAC肿瘤细胞缺乏细胞角蛋白-19的表达,细胞表面MHC-1表达降低;而在肝转移灶内MHC-I阴性的PDAC细胞中,自噬相关基因表达非常丰富。PDAC肿瘤细胞表面的MHC-I经NBR1介导的自噬-溶酶体途径减少[26],因此可以通过抑制自噬来恢复癌细胞表面的MHC-I表达水平,从而增强抗肿瘤T淋巴细胞反应,抑制肿瘤生长。
铁死亡是一种铁依赖的非凋亡性细胞死亡形式,其敏感性可作为肿瘤细胞抵抗死亡,适应性生长的机制。KRAS突变型PDAC细胞对铁死亡诱导较敏感,铁死亡的PDAC细胞可通过自噬介导的外泌体分泌释放KRASG12D蛋白,引起巨噬细胞向M2型极化并分泌免疫抑制因子,促进PDAC的发生和进展[28]。表 1总结了KRAS突变对PDAC的全方位影响,特别是代谢重编程、巨胞饮与自噬作用,有助于理解这一关键信号通路对PDAC癌细胞侵袭、转移、耐药特性的作用。
PDAC肿瘤微环境具有高度异质性,包含肿瘤细胞、细胞外基质和基质细胞。肿瘤细胞被由胶原网组成的增生组织紧密包绕,形成缺氧、营养匮乏的恶劣环境。
CAF是促进肿瘤增殖、迁移、侵袭和转移的关键因素,它主要来源于胰腺星状细胞(pancreatic stellate cells, PSC),受到KRAS突变影响[29]。CAF能够调节肿瘤免疫微环境,影响抗肿瘤药物的作用;可在激活后分泌细胞外基质蛋白(如胶原蛋白、纤连蛋白和层粘连蛋白)到肿瘤微环境中;具有促结缔组织增生作用。研究[30-31]表明,在癌变过程中,CAF可产生炎性介质如CXCL8和IL-6,介导炎症、肿瘤生长和血管生成,而KRAS/CXCR2信号通路在调节PDAC的CAF中起主要作用。
突变型KRAS对肿瘤细胞周围基质也有影响。在PDAC中,豪猪(Hedgehog)信号通路被高度激活。SHh是豪猪信号通路的配体,通过NF-κB通路和KRAS可增加SHh的分泌,从而上调多种细胞外基质成分,如胶原蛋白、基质金属蛋白酶、原纤维蛋白-1;SHh还能通过上调胰岛素样生长因子和生长停滞特异性基因6的表达来影响PSC细胞间信号传导。PSC与肿瘤细胞间的通讯可促进局部肿瘤生长和癌症的远处转移,当PDAC进展时,SHh在细胞通讯中的作用使得胰岛素样生长因子和生长停滞特异性基因6表达不断升高,最终导致肿瘤细胞增殖活跃,同时增强抵抗凋亡的作用[32]。
携带KRAS突变的PDAC细胞可分泌趋化因子如GM-CSF、IL-6。这些趋化因子激活多种免疫细胞,包括T淋巴细胞、B淋巴细胞、髓源性抑制细胞和巨噬细胞,导致肿瘤微环境的炎性状态[10]。致癌突变型KRAS刺激血管生成因子(如VEGF)的释放,激活YAP-TAZ途径及其下游Janus激酶信号转导和JAK-STAT3信号转导,影响肿瘤微环境免疫抑制的情况、远处转移的可能性和对抗肿瘤药物治疗的反应。
炎症、耐药、纤维化、血管生成等微环境改变促使PDAC发生变异并选择变异优势克隆。KRAS作为PDAC细胞突变的主要类型,该信号通路的激活增强了细胞通讯能力、提高了趋化因子和血管生成因子的分泌水平,增强了PDAC细胞适应炎性环境、免疫抑制、远处转移和耐药能力。
作为胰腺癌发生的中心,KRAS是一个关键的治疗靶点。针对KRAS的治疗策略共有3个方向:(1)直接靶向KRAS蛋白;(2)通过蛋白定位间接靶向KRAS;(3)通过干扰下游效应蛋白和信号通路影响KRAS致癌活性。但目前针对以上3方向尚无有效的药物。
KRAS的点突变包括多种情况,其中在药物治疗态势最好的是G12C突变型。AMG510、MRTX849、ARS-3248/JNJ-74699157和LY3499446均为靶向拮抗携带KRASG12C突变的PDAC细胞的药物[33]。其中AMG510和MRTX849被确认可以在携带KRASG12C突变的PDAC患者中显著缩小肿瘤大小[34-36]。此外,在携带KRASG12C突变的小鼠模型中观察到使用AMG510后出现了以CD8+为主的T淋巴细胞在肿瘤内的浸润增殖,表明AMG510对肿瘤免疫微环境的改善[34]。然而在所有PDAC的病例中,KRASG12C突变仅占不到3%的比例,而将近50%的病例是携带KRASG12D突变。目前,CD8+T淋巴细胞的过继性移植在治疗携带KRASG12D突变的结直肠腺癌患者中疗效较好,而对于大部分携带KRASG12D突变的PDAC患者,这一治疗手段仍待临床试验评估。
通过改变蛋白定位间接靶向KRAS基因的治疗方法包括异戊二烯化抑制剂、Icmt抑制剂、PDE6δ抑制剂。法尼基转移酶(FTase)介导KRAS细胞膜锚定域末端CAAX序列中半胱氨酸异戊二烯化(即法尼基化),在其中添加疏水链有利于膜稳定。法尼基转移酶抑制剂可以靶向这一修饰作用,抑制KRAS与细胞膜的稳定结合。竞争性FTase抑制剂Tipifarnib已进入胰腺癌治疗的三期临床试验,但由于GGTase介导的代替性异戊二烯化作用的存在,治疗并未起效,FTase抑制剂与GGTase抑制剂的联合使用也因为高毒性而禁用于临床治疗[37]。PDE6δ抑制剂Deltarasin是较有前景的药物,PDE6δ参与KRAS膜定位的过程,抑制剂Deltarasin可阻断KRAS突变型胰腺癌细胞上的膜定位作用,降低细胞增殖、促进细胞凋亡。
针对KRAS下游信号通路的靶向治疗中,RAF-MEK-ERK MAPK通路是KRAS下游最具特征性的通路之一,作为KRAS突变型肿瘤的治疗靶点,针对这条通路的研究广泛且深入[38]。活化的KRAS通过诱导RAF的磷酸化和二聚化激活RAF,继而启动下游环环相扣的磷酸化过程,最终ERK激活促进细胞增殖的转录因子[39]。KRAS下游的MAPK通路激活对其致癌信号至关重要。目前开发的RAF、MEK和ERK抑制剂中,RAF抑制剂vemurafenib和dabrafenib对KRAS突变型恶性肿瘤治疗失败。
由KRAS信号激活的PI3K-AKT-mTOR通路也是药物开发关注的中心通路,针对PI3K通路的抑制剂用作单一药物治疗PDAC疗效不佳,推测原因是KRAS下游的信号通路存在交互,即使抑制了单一通路的某一环节也可“变道”至其他通路激活。因此,同时靶向PI3K和MAPK通路的联合治疗成为目前的潜在策略。
PDAC的发病率呈逐年上升趋势,其5年生存率极低,病死率与发病率几乎接近。这归咎于其高侵袭性及转移性,而高KRAS突变率被认为是PDAC起始及进展的主要驱动因素。显而易见,KRAS突变除刺激癌细胞分化、增殖、黏附、迁移,直接导致高侵袭性和转移性外,还控制糖、脂、氨基酸及能量代谢重编程,使其适应缺氧、炎症、免疫抑制及纤维化等微环境改变,逃逸免疫攻击。KRAS信号通路无疑成为PDAC治疗的主要靶标,但众多临床试验结果并不满意。新KRASG12C抑制剂只能使一小部分PDAC患者获益,而对于PDAC的主要突变型KRASG12D目前并没有批准的药物。分子靶向治疗结合免疫治疗给PDAC患者带来新希望,临床试验也正在积极研究多种KRAS相关途径和分子,寻找可能的治疗靶点。目前正在开展联合疗法的临床试验有:检查点抑制剂Pembrolizumab联合KRASG12C抑制剂Sotorasib(NCT04185883)、Pembrolizumab联合Adagrasib(NCT04613596)、Sintilimab联合LY3537982(NCT04956640)、Atezolizumab联合GDC-6036(NCT04449874)、Spartalizumab联合JDQ443(NCT04699188)[40]。此外,考虑到PDAC特殊而复杂的肿瘤微环境,将KRAS靶向药物与胰腺肿瘤微环境调节剂相结合,提高药物递送效率从而增强疗效,这一思路也有待临床试验验证其疗效及安全性。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017