









200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




李 知 张 倜
DOI:10.3969/j.issn.1006-298X.2025.06.017
[基金项目]国家自然科学基金青年项目(82300770);东部战区总医院基础研究计划(2023JCYJYB125)
[作者单位]东部战区总医院 国家肾脏疾病临床医学研究中心(南京,210016)
[通信作者]张 倜(E-mail:drteezhang@163.com)
摘 要 33岁女性患者,因“发现尿检异常20年,大量蛋白尿4月”入院。患者20年前因神经系统异常伴少量蛋白尿于外院诊断为Wilson病,长期予青霉胺驱铜治疗。本次因自行停用驱铜药一年余后Wilson病复发,肾损伤从少量蛋白尿,逐渐发展为肾病综合征,血清肌酐正常。完善肾活检检查,病理示肾小球膜性病变,免疫荧光染色示IgG4、IgA、C3阳性为主,抗磷脂酶A2受体、含血小板反应蛋白1型结构域7A蛋白等膜性肾病抗体均阴性,电镜下见肾小球足细胞足突广泛融合,诊断为Wilson病相关膜性肾病。予以他克莫司治疗后患者肾病综合征达到临床缓解状态。
关键词 Wilson病 肝豆状核变性 膜性肾病
LI Zhi, ZHANG Ti
National Clinical Research Center for Kidney Diseases, Jinling Hospital, Nanjing 210016, China
Corresponding author:ZHANG Ti(E-mail:drteezhang@163.com)
ABSTRACT This is a case of a 33-year-old female patient who was admitted to our hospital due to "abnormal urine test results detected for 20 years and massive proteinuria for 4 months". Twenty years ago, the patient was diagnosed with Wilson’s disease at another hospital based on abnormal neurological manifestations accompanied by mild proteinuria, and had received long-term penicillamine therapy for copper chelation. The current admission was prompted by a relapse of Wilson’s disease after self-discontinuation of copper-chelating medication for over a year. The renal injury presented with mild proteinuria as the initial manifestation, progressed gradually to nephrotic syndrome, and the serum creatinine level was within the normal range. Renal biopsy revealed membranous nephropathy, with immunofluorescence staining predominantly positive for IgG4, IgA, and C3, while antibodies associated with membranous nephropathy such as anti-PLA2R and anti-THSD7A were negative. Electron microscopy demonstrated extensive effacement of podocyte foot processes. A diagnosis of Wilson’s disease-associated membranous nephropathy was established. The patient achieved clinical remission of nephrotic syndrome following treatment with tacrolimus.
Key words Wilson disease hepatolenticular degeneration membranous nephropathy
33岁女性患者,因“发现尿检异常20年,大量蛋白尿4个月”入院。患者2003年4月无诱因出现双下肢轻度水肿及头部不自主震颤表现,至当地医院就诊查尿蛋白+/-,尿蛋白定量0.34 g/24h,血清肌酐(SCr)113.8 μmol/L,进一步至安徽中医药大学神经病学研究所附属医院完善相关检查后诊断“Wilson病”(病历资料及检查报告未见),予口服青霉胺规律驱铜治疗(0.5 g/d)至2021年自行停药,自诉期间未再出现水肿及头部震颤等不适,定期复查尿蛋白持续阴性(未见报告)。2022年4月患者无诱因出现双手及头部不自主抖动,再次至安徽中医药大学神经病学研究所附属医院就诊。
查体示四肢肌力V级,右上肢腕关节肌张力轻度铅管样增高,其余三肢体肌张力减低,双上肢轻中度姿位性震颤,头部轻度震颤,双侧巴宾斯基征阴性,双手快复轮替动作笨拙,指鼻试验欠稳准,全身无水肿。
实验室检查尿蛋白+,尿红细胞98.56/μL,SCr 57 μmo1/L,血清白蛋白(Alb)38.1g/L,谷丙转氨酶15U/L,谷草转氨酶19 U/L,血红蛋白137 g/L,血小板114×109/L,铜蓝蛋白62.4 mg/L(正常值200~400 mg/L)。
头颅MRI:双侧基底节区异常信号,符合肝豆状核变性MR表现;肝胆胰脾超声:肝豆肝病样改变(结节型)、肝脏中度纤维化;裂隙灯下见角膜 Kayser-Fleischer(K-F)环++++。
诊断“肝豆状核变性(脑-内脏型 Ⅱ级)、肝硬化、蛋白尿”,予二巯基丙磺酸钠(DMPS)(0.75 g/d,1~6 d/周×13周)驱铜治疗,治疗期间监测24 h尿铜1 056.05~2 907.77 μg/24h(正常值<100 μg/24h),尿蛋白定量3.1~3.9 g/24h,Alb 20.2~23.4 g/L。
2022年9月出院后继续口服二巯基丁二酸(DMSA)0.25 g 2次/d×4天驱铜治疗。2022-11-25因大量蛋白尿持续不缓解至肾脏疾病临床医学研究中心就诊,查尿蛋白定量5.45 g/24h,SCr 55.7 μmol/L,Alb 26.80 g/L,予收治入院。
患者已婚未育,近1年月经不规律,近2月无月经,既往史无特殊,父母非近亲结婚,否认家族有Wilson病等遗传病史;患者神经系统查体未见明显异常,肉眼观察未见特征性角膜K-F环,双下肢对称性轻度水肿。
体质指数37.18 kg/m2,尿蛋白定量3.44 g/24h,尿红细胞计数56.2/μL,血常规白细胞计数2.35×109/L,淋巴细胞计数1.00×109/L,血红蛋白浓度124 g/L,血小板计数85×109/L;SCr 56.6 μmol/L,Alb 23.90 g/L,前白蛋白122 mg/L,血清总蛋白48.20 g/L,估算的肾小球滤过率(eGFR) 118 mL/(min·1.73m2),铜1.9 μmol/L,钙1.80 mmol/L,钾3.31 mmol/L,钠145.1 mmol/L,氯110.8 mmol/L,总胆固醇6.88 mmol/L,凝血功能未见明显异常,糖化血红蛋白(HbA1c)5.4%。抗磷脂酶A2受体(aPLA2R)抗体<2.00 RU/mL;抗核抗体滴度1∶512,抗双链DNA抗体<1∶10,抗核抗体谱阴性;补体C3/C4、免疫球蛋白、淋巴细胞亚群、免疫固定电泳图谱及血游离轻链比值未见异常;传染病四项阴性。
肾脏超声:双肾皮质回声增强(109 mm/102 mm);全腹部CT平扫:肝脏体积缩小,肝脏边缘形态不规整,呈波浪状改变;肝实质密度正常,未见明显异常密度区。
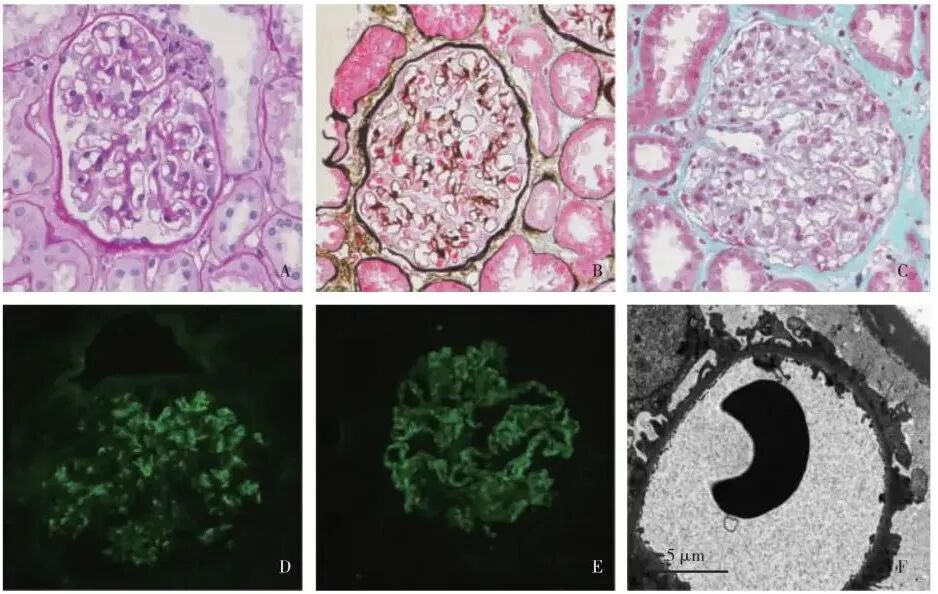
肾活检(2023-02-20)光镜下见31个肾小球中1个球性废弃,余肾小球毛细血管袢开放好。节段系膜区轻度增宽,囊壁节段增厚(图1A)。PASM-Masson染色示肾小球上皮侧少量嗜复红物沉积(图1B)。肾小管间质轻度急性病变(图1C)。动脉未见明确病变。免疫荧光见IgG++、IgA++(图1D)、κ轻链+、λ轻链++ 呈颗粒状节段沉积于系膜区及血管袢。C3++、C1q+、IgG1+、IgG3 trace、IgG4++(图1E)呈颗粒状弥漫沉积于系膜区及血管袢;IgM trace呈颗粒状节段沉积于系膜区。IgG2、PLA2R、Fibrin阴性;α3 、α5链表达正常。免疫组化示肾组织神经表皮生长因子样蛋白1、外生骨疣蛋白1、外生骨疣蛋白2、信号素3B、原钙黏蛋白7、含血小板反应蛋白1型结构域7A蛋白均阴性;电镜下观察到2个肾小球,毛细血管袢开放好,基膜普遍偏薄,140~250 nm,较厚处约380 nm,节段基膜上皮侧高密度的电子致密物沉积(图1E),个别致密物周围见钉突形成,基膜内皮下未见电子致密物沉积。节段系膜区增宽,系膜基质增多,系膜区亦见较多电子致密物沉积。肾小球足细胞足突广泛融合(80%~90%),胞质内见吞噬溶酶体。患者及家属拒绝行基因检测。
经排除感染、自身免疫性疾病、肝炎、肿瘤及药物等致病因素后,最后诊断:(1)膜性肾病(Wilson病相关);(2)慢性肾脏病1期;(3)Wilson病(脑-内脏型 Ⅱ级);(4)肝硬化;(5)白细胞减少;(6)血小板减少;(7)肥胖症。患者出院后继续服用二巯基丁二酸(1 g,2次/d)驱铜治疗,同时予他克莫司(1 mg, 1次/12h)、缬沙坦胶囊(80 mg,1次/d)及升白细胞、升血小板、护肝等治疗。
2023-06-28门诊复诊:白细胞计数4.65×109/L,血小板计数99×109/L,尿蛋白定量0.53 g/24h,SCr 55.7 μmol/L,Alb 37.7g/L,复查血aPLA2R抗体阴性,他克莫司谷浓度5.3 ng/mL,维持他克莫司治疗。2023年12月复诊尿蛋白维持阴性。2024年5月自然受孕,停用他克莫司,定期孕检蛋白尿维持阴性。2025年1月经剖宫产诞下1健康男婴(35周+4 d),婴儿未行基因检测。2025年6月于安徽中医药大学神经病学研究所附属医院行定期DMPS驱铜治疗,其间复查蛋白尿阴性,SCr正常。
图1 A:肾小球节段系膜区轻度增宽,囊壁节段增厚(PAS,×400); B: 肾小球上皮侧少量嗜复红物沉积(PASM-Masson,×400); C:肾小管间质轻度急性病变,间质散在单个核细胞浸润(Masson三色,×400); D: IgA++,呈颗粒状沉积于系膜区及血管袢(IF,×400); E: IgG4++,呈颗粒状沉积于血管袢及系膜区(IF,×400); F:肾小球节段基膜上皮侧高密度的电子致密物沉积(EM)
本例为目前公开文献中首次报道继发于Wilson病的膜性肾病,其肾脏病理以肾小球膜性病变为主,但同时伴系膜区增宽、系膜区电子致密物沉积及IgA、C3沉积等典型IgA肾病病理表现,以单纯膜性肾病作为诊断是否合适,经与病理医生讨论,该患者肾脏病理已达到IgA肾病诊断标准,但其病理表现以肾小球膜性病变为主,临床表现为肾病综合征,最终仍考虑诊断为膜性肾病。临床上膜性肾病合并IgA肾病并不鲜见,一项研究总结了26例膜性肾病合并IgA肾病患者的临床和病理特征,结果显示膜性肾病合并IgA肾病患者与单纯膜性肾病患者具有相似的临床特征,并且比单纯IgA肾病患者的病理损害更轻[1],另一项涵盖137例患者的回顾性队列研究显示,膜性肾病合并IgA肾病患者与单纯膜性肾病患者的临床和病理特征更相似,建议此类患者以膜性肾病为诊断并进行治疗[2]。所以本例患者诊断为膜性肾病。
膜性肾病是一种抗体介导的自身免疫性肾小球疾病,是成人肾病综合征中最常见的病理类型[3-4]。Hou等[5]报道膜性肾病在中国的慢性肾小球肾炎中约占23.4%,仅次于IgA肾病。膜性肾病根据其病因可分为原发性和继发,75%~80%的患者在缺乏基础病因的情况下发生,称为原发性膜性肾病,其余病例则继发于一些明确的病因,称之为继发性膜性肾病[3]。近年来关于原发性膜性肾病的重要进展在于PLA2R等膜性肾病相关自身抗原的发现,从分子层面阐释膜性肾病的发病机制[6]。而基础病因的发现则对继发性膜性肾病至关重要,在控制基础病因的基础上,治疗通常可以取得良好效果。目前已知包括系统性红斑狼疮等自身免疫性疾病、感染、肝炎病毒(如乙型病毒性肝炎)、药物、肿瘤等均可诱发膜性肾病[3]。目前已有继发于金属汞的膜性肾病的病例报道,如Gao等[7]收治172例汞中毒患者,对其中35例肾病综合征患者行肾活检,18例诊断为膜性肾病。而继发于铜的膜性肾病还未见报道。
Wilson病,又称为肝豆状核变性,是一种遗传性铜代谢障碍性疾病,因编码跨膜铜转运三磷酸腺苷(ATP)酶的ATP7B基因突变导致铜在肝、脑、肾等器官病理性蓄积而致病[8]。据报道我国汉族人群Wilson病的发病率和患病率分别约为1.96/10 000和5.87/10 000[9]。人体主要通过饮食摄入铜,在肠道中由肠上皮细胞摄取后大部分被肝脏吸收,后经胆汁由消化道排泄。体内铜的稳态主要通过调节小肠中铜的摄取和胆汁中铜的排泄这一动态平衡来实现。当ATP7B基因突变时,铜因排泄障碍而在肝脏沉积,当超过肝脏储存上限后以游离铜的形式进入血液,并在脑部、肾脏、角膜、关节及肠道等部位沉积,引起相应的临床表现[8,10]。肝脏的ATP7B基因表达最高,是调节全身铜稳态的中心器官,所以肝损伤是Wilson病最早和最常见的临床表现[11]。其次是神经系统症状,表现为肌张力障碍、震颤等[12];其他典型体征还包括角膜K-F环,骨关节功能障碍及肾功能损伤等[13]。本例患者存在神经系统症状、脑部MRI改变、肝脏受损、眼部K-F环、蛋白尿等多系统损伤表现,根据2001年莱比锡第8届Wilson病国际会议制定的诊断标准(Leipzig评分系统)[14],Wilson病诊断明确,但继发膜性肾病的致病机制还需要进一步探讨。
Wilson病相关肾损伤早有报道,一项研究显示,相比正常人群,Wilson病特别是合并神经系统损伤的患者SCr和尿素氮水平相对较高[15]。Dang等[16]综述了肾小管铜蓄积所致的肾小管功能障碍,包含从轻度的水电解质和酸碱平衡紊乱到范可尼综合征,近端和远端肾小管酸中毒促进肾结石、肾钙质沉着和骨代谢异常的发展,间接并发症包括肝肾综合征或心肾综合征导致的肾灌注不足,甚至急性溶血、横纹肌溶解或胆汁型肾病等,同时其提出Wilson相关肾小球损伤主要与驱铜药物青霉胺的药物毒性有关。而Zhuang等[17]回顾分析85例Wilson病患儿,排除9例接受青霉胺治疗者后,有14例同时出现蛋白尿和血尿。Jin等 [18]统计了20例Wilson病合并肾损伤患者病例资料,临床表现以肉眼血尿及水肿为主,8例患者24 h尿蛋白定量增加,6例在0.15~3.5 g/24h,2例>3.5 g/24h。本例患者20年前起病即有少量蛋白尿,本次Wilson病复发前已停用青霉胺1年余,复发时肾损伤表现为肾病综合征。而本中心2010年报道的初发Wilson病例,其肾损伤表现为少量蛋白尿伴镜下血尿,同时存在低尿酸血症、低钾血症,提示近端肾小管功能损害[19]。综上可见Wilson病相关肾损伤的临床表现异质性较大,可能与铜在肾脏沉积的部位及蓄积量有关。
Wilson病合并肝脏及神经系统损伤更为常见,患者通常由肝病科及神经内科首诊,同时因肝损后凝血异常及精神异常等因素,Wilson病合并肾损伤后行肾活检的病例不多见。我中心2010年报道病例经肾活检诊断为继发性IgA肾病[19]。Jin等[18]收集7例完善肾活检病例,其中3例为IgA肾病,1例为微小病变性肾病,2例为膜增生性肾病,1例为局灶节段性肾小球硬化。结合本病例,似乎继发于Wilson病的肾病病理类型以IgA肾病多见。因Wilson病常伴肝损伤,肝脏清除循环中IgA能力的下降,致病性IgA在系膜区的沉积增多可作为部分解释。其他病理类型如本例膜性肾病及膜增生性肾病、局灶节段性肾小球硬化等可见多种免疫复合物(IgA、IgM、IgG、补体C3、补体C1q等)沉积于多部位(系膜区、基膜、毛细血管壁等),提示铜可于肾小球多部位沉积后形成免疫复合物,继而激活补体系统引起肾脏免疫损伤。而微小病变性肾病及局灶节段性肾小球硬化等伴寡免疫复合物的病例,提示铜可能直接在足细胞沉积后诱发足细胞损伤,破坏肾小球滤过屏障,继而产生大量蛋白尿,但损伤机制未知。
本病例局限在于缺乏铜沉积肾组织的直接证据。既往有报道用组织化学方法进行铜染色,包括罗丹宁、地衣红、红氨酸和Timm法等[14],但本中心未开展相应技术。笔者曾尝试用本例患者的肾组织切片进行电镜下扫描能谱仪的成分分析,但未发现铜元素,分析可能与病理切片部位、扫描仪扫描阈值等因素有关,后续待增加样本量后进一步尝试。
根据一项涉及3 285例老年人的横断面研究,血铜水平与慢性肾脏病患病率呈剂量依赖性正相关[20]。目前通过动物模型的探究,初步发现,铜可通过氧化应激、氧化还原状态、能量代谢障碍、线粒体功能障碍等方面引起肾脏损伤[21-24]。2022年Tsvetkov等提出铜死亡概念[25],其区别于目前已知的凋亡、焦亡、坏死、铁死亡等细胞死亡方式,机制为细胞内铜过载时,其结合三羧酸循环中的脂酰化蛋白,导致铁硫簇蛋白失活及硫辛酰化蛋白的聚集,从而引发细胞死亡,该概念为探索Wilson病及铜相关肾损伤的致病机制提出新方向。
回顾本例患者,20年前经外院诊断Wilson病,予以长期青霉胺驱铜治疗后病情呈稳定状态。1年前自行停药后本次再发Wilson病伴肾病综合征,予DMPS、二巯基丁二酸(DMSA)等序贯驱铜治疗,同时至国家肾脏疾病临床医学中心完善肾活检后病理明确为肾小球膜性病变为主。此次病情再发前,患者已停用青霉胺1年余,而本次使用的排铜药物DMPS、DMSA,经PubMed、知网等文献检索平台检索,无相关肾损伤报道。排除药物因素后,患者诊断为膜性肾病(Wilson病相关),经他克莫司单药治疗后患者肾病综合征缓解,尿蛋白转阴。但相关致病机制需要进一步探索。
小结:Wilson病即ATP7B基因突变导致的铜异常代谢,可致铜于肾脏多部位(肾小球基膜、系膜区、足细胞及肾小管等)沉积并造成肾损伤。本文报道的1例继发于Wilson病的膜性肾病,使用他克莫司治疗后尿蛋白转阴。但铜相关肾损伤的具体机制仍需进一步研究。在定期排铜的基础治疗下,定期完善尿检、监测肾功能可尽早识别Wilson病相关肾损伤,同时建议有条件者完善肾活检明确病理类型。
参考文献
【引用本文】李知、张倜. Wilson 病相关膜性肾病[J]. 肾脏病与透析肾移植杂志, 2025, 34(6): 590-594.
LI Zhi, ZHANG Ti. Wilson’s disease⁃associated membranous nephropathy: a case report[J]. Chinese Journal of Nephrology, Dialysis & Transplantation, 2025, 34(6): 590-594.
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017