










200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:复旦大学附属华山医院神经内科 王宁宁、祝搏晨
审核:复旦大学附属华山医院神经内科 朱雯华
本次世界肌病学会年会(WMS),多项关于脊髓性肌萎缩症 (SMA) 的研究展示了过去一年在早期干预疗效、全身性病理机制解析、非运动神经元系统损伤以及高危人群筛查等方面的全链条创新成果。这些研究不仅为理解疾病机制提供了新的视角,特别是对SMA这一全身性疾病的认识深化,更为临床试验设计和未来联合治疗方案的开发奠定了坚实基础。以下是本次会议中SMA领域的重要研究亮点。
SMA真实世界治疗疗效
诺西那生钠作为一种反义寡核苷酸,可修饰SMN2基因的剪接以增加中枢功能性SMN蛋白的生成。NURTURE研究是一项2期单臂、开放标签研究,旨在评估在症状出现前开始使用诺西那生,对症状前SMA患儿的安全性和有效性。
研究设计和主要研究结果
纳入的受试者均经基因确诊但尚未发病的症状前SMA,首次给药时年龄≤6周,根据SMN2拷贝数,分为3个亚组,包括3拷贝组,2拷贝组和2拷贝亚组(基线腓神经CMAP波幅≥2mV,且体格检查腱反射阳性)。共25名患儿纳入研究,从2015年开始随访7.9 (5.9–8.3) 年。启动诺西那生治疗后,基线NfL水平升高的患儿其NfL水平快速下降,基线在正常范围内的患儿,其NfL水平可以保持低位,且该效应随时间推移持续存在,表明神经退行性变被持续抑制。研究结束时,所有受试者均存活,80%的患者不需要呼吸干预治疗,大多数患者都能在正常发育时间中达到运动功能里程碑,HFMSE和6MWT运动功能评分随着时间的增加明显改善。安全性数据显示总体耐受较好,8年随访期间未出现新的安全性事件,腰椎穿刺耐受良好。在症状前阶段启动诺西那生治疗患儿,其临床结局优于未治疗或治疗较晚的患儿(具有相似SMN2拷贝数)。
利司扑兰作为SMN2剪接修饰剂,与诺西那生钠不同的是,通过口服给药能够增加整个中枢神经系统和机体外周组织的SMN蛋白水平。RAINBOWFISH研究是一项多中心、开放标签、单臂研究,旨在评估利司扑兰在已基因确诊但在基线时没有出现临床症状或体征的SMA患儿中的疗效和安全性。
研究设计和研究结果
主要终点为BSID-Ⅲ量表评估12个月时能够独坐的SMA患儿比例。次要终点包括SMA临床症状的进展情况、生存和永久通气、运动里程碑的达成、CHOP-INTEND量表、吞咽和经口进食能力、住院情况。此外,该研究还有两个探索性终点:通过BSID-Ⅲ认知量表评估的患儿认知功能以及通过神经系统检查评估语言发育情况。共有23例患儿完成了为期3年的治疗,3例患儿中途退出,根据BSID-Ⅲ和HINE-2,治疗3年后,91%(21/23)的患儿都能独坐、独站和行走。96% (22/23)均能够完全经口进食,且不需要机械通气,所有患儿语言发育功能和吞咽功能正常,认知能力表现出与正常儿童发育一致的水平。85%的患儿在整个研究期间未曾住院。
安全性数据显示耐受良好,常见的不良反应包括胃肠道症状、肺部感染等,治疗期间未出现导致停药或剂量调整的治疗相关不良事件。
SMA治疗方法讨论及新治疗方法
SMA治疗:局部治疗 OR 全身性治疗?
来自University of Freiburg的 Kirschner教授深入探讨了SMA作为一种全身性疾病的特性,并比较了局部与全身性治疗策略的研究现状。首先概述了SMA的临床症状和发病机制,强调了SMN2拷贝数与疾病严重程度之间的明确关联。他指出,SMN蛋白在人体内普遍表达,但其水平受发育调控,在胚胎期和新生儿期达到高峰。然后讲者介绍了SMA非运动神经元受累现象在研究中的挑战:1、临床方面:难以区分患者的原发性受累与继发性变化,以及判断症状是否与SMN相关的病理有关;2、动物模型:SMN减少的表现存在组织特异性,且动物模型的研究结果并非总能成功转化到人类。3、临床试验:主要挑战包括样本量小和观察期短。在治疗方面,目前主要有两个方向:基因治疗(补充SMN1)和小分子药物(增加SMN2表达)。基于疾病的全身性本质,教授提出了一个关键问题:当前治疗是否应该超越单一地针对运动神经元,而拓展至全身系统?为了回答这一问题,讲者介绍了SMArtCARE真实世界治疗结果。数据显示,一旦患者出现症状,其行走能力会迅速丧失,突显了早期干预的必要性。基于对超过5000例不良事件的分析,研究发现:在比较局部(针对中枢神经系统-CNS-targeted)治疗与全身性治疗时,未观察到其他器官受累的聚集现象。这意味着局部 CNS 靶向治疗的安全性风险模式与全身性治疗相似,全身性治疗的额外益处尚未得到明确证实。最后Kirschner教授总结强调由于SMN表达的时间特异性,早期启动治疗至关重要。然而,某些症状(如神经发育问题)出现得极早,目前尚不明确后续的补充性治疗能否改善这些在产前或围产期已经出现的病理表现。
SMA新生儿筛查能够早期识别SMA,改善治疗预后,在德国和澳大利亚已得到广泛实施。筛查中被NGS识别出两名SMN1缺失新生儿,分别来自德国(P1)和澳大利亚(P2)。P1无SMA家族史,来自德国的患者1后续遗传学验证分析确定了复合杂合变异,即SMN1第7外显子大缺失和第7外显子内4个碱基对缺失【c.855_858delp.(Arg288Alafs*5)】,同时伴随SMN2拷贝数为零,来自澳大利亚的患者2遗传学验证分析为SMN1第7外显子大片段缺失和第7外显子内4个碱基对缺失【c.861_864del(p.Arg288Alafs*5)】,SMN2拷贝数为1。
建立斑马鱼模型研究两个变异型的致病性,结果显示虽然两名SMN蛋白水平低,热稳定性不受影响,提示表明变异产生的SMN蛋白结构稳定,最后决定不治疗和等待观察。两个患者都没有症状,显示出正常的运动、神经和电生理检查结果。两个患者在预期时间达到了所有运动里程碑,都避免了不必要的治疗和相应的潜在副作用和高昂的治疗费用。尽管全长SMN完全缺失,但极低水平的新型SMN蛋白仍保持正常的运动功能。
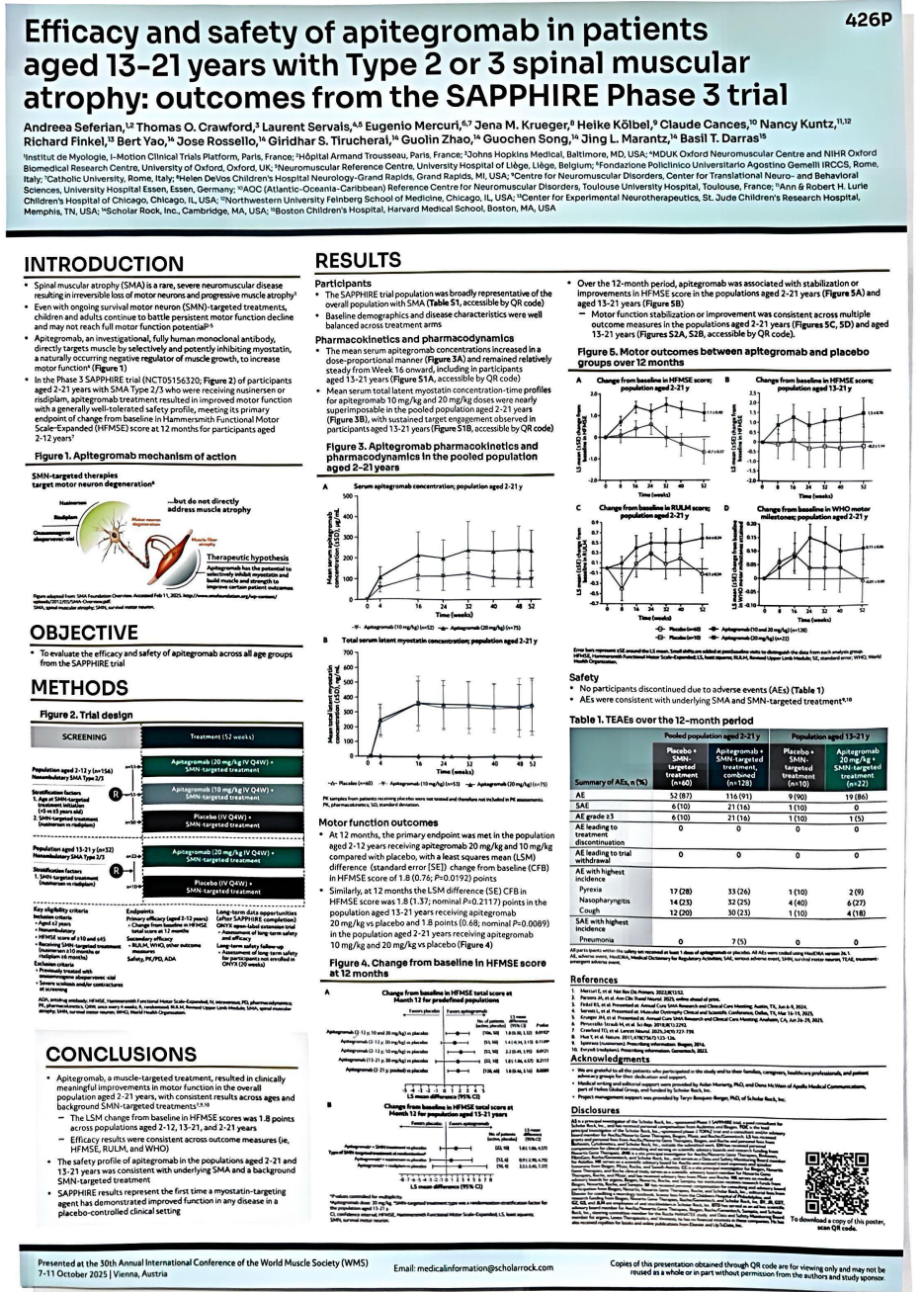
目前批准的SMA疗法主要通过增加SMN蛋白表达减缓疾病进展,但这些疗法主要针对运动神经元退化,并未解决伴随的肌肉萎缩问题。Apitegromab是一种全人源化单克隆抗体,通过抑制肌抑素激活来改善肌肉功能。一项针对Apitegromab随机、双盲、安慰剂对照的3期临床试验(NCT05156320),旨在评估Apitegromab在2型和3型且丧失行走能力SMA患者的安全性和疗效,且入组患者正在接受诺西那生钠或利司扑兰治疗。
研究设计和主要数据
2-12岁的患者按1:1:1随机分配至接受Apitegromab 20mg/kg、10mg/kg或安慰剂组,13-21岁的受试者按2:1随机分配至接受Apitegromab 20mg/kg或安慰剂,皆每4周静脉输注(IV)一次治疗,治疗疗程最长持续12个月(52周)。研究共纳入了188例SMA 2型或3型患者,这些患者基线HFMSE评分在10-45分之间,并且在筛选时已经接受了诺西那生或利司扑兰治疗。
与安慰剂组(n=50)相比,2~12周岁患者中Apitegromab组(n=106)的HFMSE评分的平均变化(vs基线)增加1.8分。13~21周岁患者疗效评估分析显示类似结果。尽管所有研究患者都在接受SMN靶向治疗,但与安慰剂相比,接受Apitegromab治疗的SMA患者在52周时,表现出持续的运动功能评分改善。从安全性数据显示受试者对Apitegromab治疗耐受良好,没有因药物引起的不良事件导致治疗的中断。该研究结果支持肌肉靶向治疗在改善SMA患者运动功能方面的潜力,尤其是在SMN靶向治疗无法解决的肌肉萎缩问题上,为SMA患者带来新的曙光。
来自Newcastle University的Jordi教授在演讲中深入分析了SMA和肌萎缩侧索硬化症(ALS)患者肌肉组织中的分子变化,提出去神经支配的肌肉并非被动受害者,而是积极参与再神经支配过程。通过对SMA肌肉进行单细胞和snATAC测序,其团队发现了一系列关键的分子证据:一方面,肌核中血管发育相关基因上调,以及参与神经肌肉接头发育的基因上调,这表明肌纤维正在主动重塑并尝试形成新的神经肌肉接头(即再神经支配的努力);另一方面,参与神经元发育的基因、RNA加工、核糖体功能和线粒体功能的基因均有减少,反映了肌肉细胞基础功能和代谢的受损。进一步对比发现,ALS患者的细胞群体也发生了类似的变化,表明这两种运动神经元疾病在肌肉病理层面共享分子机制,尽管ALS的肌纤维损失更明显,且两种疾病的基因表达仍存在诸多差异。最终,教授总结指出,正是这种肌肉的主动修复和重塑机制,为未来针对肌肉-神经交互作用的运动神经元疾病(MNDs)联合疗法,带来了巨大的潜力。
SMA非运动神经元系统损伤
SMA患者中枢神经系统非运动区域的变化及其带来的认知问题
来自University Hospital Essen的Kölbel教授分享了SMA患者中枢神经系统非运动区域的变化及其带来的认知问题。首先已有的神经病理学证据表明SMN蛋白不仅对运动神经元存活至关重要,也在前脑的不同区域发挥重要的发育作用。目前,针对通过新生儿筛查(NBS)确诊的学龄前SMA患者的认知发展,研究仍存在巨大空白,尤其是未经治疗前SMA1型患者的认知状况更是未知。此外,产前SMN蛋白缺乏是否影响儿童认知功能仍不明确。为了调查这一问题,研究人员对通过 NBS确诊并接受早期治疗的患者群体进行了 BSID-III认知测试,发现了SMN2拷贝数与认知发展得分呈负相关。这表明了认知受损可能与产前或极早期发育阶段的遗传缺陷相关。其团队进一步收集和分析患者的脑脊液样本,发现神经粒蛋白(Neurogranin)可能是一个潜在的biomarker来监测患者的认知问题,但研究仍处于初步阶段,需要更大的样本量来确认这些生物标记物和认知/治疗反应之间的关联。
SMA患者的肾脏损害
SMA虽然是运动神经元退行性疾病,但也累及到多器官,此研究聚焦于SMA患者的肾脏损害以及治疗对肾脏功能的影响。
共263名SMA患者纳入分析,研究认为对于SMA患者,基于肌酐的eGFR的计算值不太可靠,基于胱抑素c的eGFR是一种有价值的替代方法。51.7%患者血钾降低。共收集83例患者的24h尿液标本,尽管血钾水平降低,但肾脏分泌的钾量正常,提示SMA患者有发生(重度)低钾血症的风险。共有231例患者接受诺西那生钠或利司扑兰治疗,随访时间分别为30个月和18个月,随访期间eGFR下降,诺西那生钠治疗组患者每年5.6ml/min/1.73m²,利司扑兰治疗患者每年4ml/min/1.73m²,治疗可部分逆转肾小管功能障碍,但eGFR甚至显著下降。
SMA高危筛查
中国多中心SMA电生理高危筛查
这项由复旦大学附属华山医院主导的中国多中心回顾性研究,旨在建立并验证一种临床-电生理联合的高风险筛查方法,以解决青少年和成人5q脊髓性肌萎缩症诊断延迟的问题。研究回顾了16万余份电生理报告,最终有1632份符合高风险筛查的电生理特征。该筛查方法对 SMA表现出100%的敏感性和85.9%的特异性。研究发现,SMA患者更倾向于15岁前发病(91.4%)和病程超过12个月(98.3%)。此外,正中神经/尺神经CMAP振幅比值≥0.865对预测SMA具有显著价值。研究建议,符合“疑似SMA”特征的患者,特别是发病年龄小于15岁且病程超过1年的患者,应优先进行SMN基因检测。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017