


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




临床上细菌感染的诊断依赖于对临床样本的培养,即在35℃左右的温度下、在特定的培养基中孵育24小时或更长时间后观察有无细菌生长。使用质谱法或根据表型特性可以对生长细菌的种属进行鉴定。将细菌暴露于一系列抗菌药物中,根据细菌的生长情况可以在表型层面评估细菌的敏感性和耐药性。
为了更快速地进行病原菌鉴定和耐药基因检测,研究者开发了基于二代测序(NGS)的新方法,称为临床宏基因组学(CMg)。临床宏基因组学即样本的宏基因组测序,其目的是在更短的时间内鉴定感染的微生物并确定它们对抗菌药物的敏感性。宏基因组学具有识别抗生素耐药基因(ARG)和发现耐药性相关突变的能力,已成为一种潜在的诊断工具。然而从细菌基因型数据推断实际表型是一个重大挑战,还有许多问题需要克服。
抗生素耐药基因数据库
2009年发布的抗生素耐药基因数据库(ARDB)试图统一分散在各个数据库中的数据,但随后没有对数据库进行维护。2012年发布了ResFinder数据库,随后在2013年发布了综合抗生素耐药性数据库(CARD)。CARD利用抗生素耐药性本体(the antibiotic resistance ontology)将ARG或耐药基因突变与耐药基因家族、作用机制、靶点和抗生素等数据联系起来。最近,国家生物技术信息中心(NCBI)发布了AMRFinder,这是一种旨在识别ARG和突变的工具。
然而,目前没有任何ARG数据库是详尽无遗的,仍有许多潜在的ARG有待确定,此外,上述数据库偏向于临床分离的病原菌,来自环境和肠道的细菌数量非常有限。对于CMg应该使用哪个数据库目前并没有达成共识。
鉴定宏基因组中抗生素耐药性的决定因素
可以使用reads或contigs两种主要方法来识别宏基因组中的ARG,contigs方法需要以给定数量的reads为基础(图1)。Contigs方法的敏感性和特异性主要取决于用于搜索ARG的参数。使用不同级别的E值时,Hmmer和BLAST产生了不同的命中数(图2)。CMg的敏感性可能受到背景宿主DNA的污染,会影响细菌及ARG的检测结果。只有当样本中细菌的基因组已完全测序时才能确保CMg的准确性,通常来说需要设定一个阈值,来确保结果的可信程度,但目前对于这一阈值暂未达成共识。
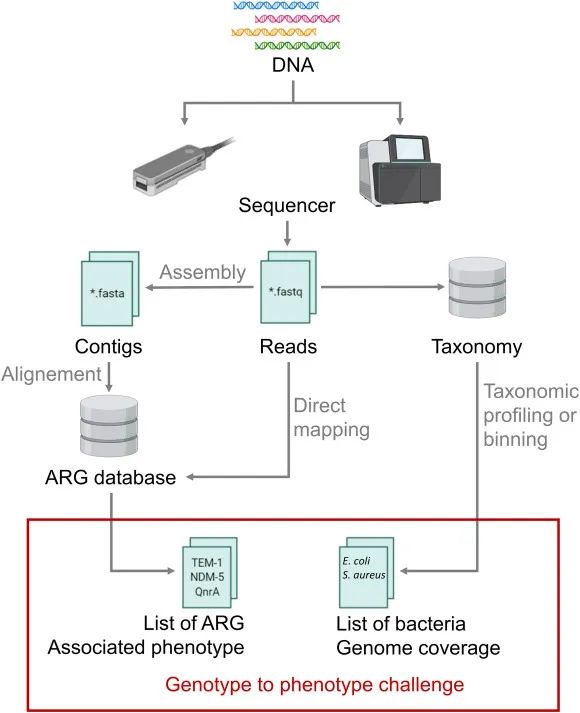
图1. 在宏基因组数据库中搜索抗生素耐药基因(ARG)的示意图。
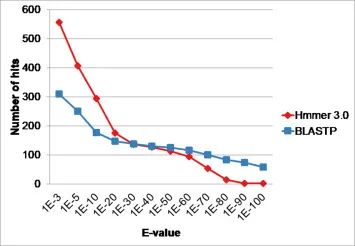
图2. 使用相同的A类β-内酰胺酶数据库和根据3.9M蛋白质目录,BLASTP和Hmmer的E值获得的命中数。(E值描述了在搜索特定的数据库时可以“预期”偶然看到的命中数)
从基因型到耐药表型
CMg获得了ARG和耐药性相关的突变信息后,一个重要的挑战是将基因型与耐药性表型相关联。对于常见的ARGs(通常是最近发现或广泛存在于病原菌中的耐药基因),它们的耐药性表型和编码的蛋白质已被广泛研究(例如编码广谱β-内酰胺酶blaCTX-M-15的基因)。然而还有一些耐药基因或突变仅在基因组数据中被鉴定到,尚无详尽描述。此外,一部分氨基酸的突变不会产生表型的改变,而另一部分突变则会明显改变细菌对抗菌药物的敏感性。研究发现金黄色葡萄球菌、大肠埃希菌、肺炎克雷伯菌、沙门菌、其他肠杆菌、屎肠球菌、粪肠球菌和空肠弯曲菌的ARGs与临床中常规使用的抗菌药物耐药性表型谱之间具有极好相关性。此外,在呼吸道标本中观察到结核分枝杆菌的基因型和表型之间也存在非常好的一致性。
尽管如此,仍有一些细菌对某种抗菌药物的耐药机制尚未完全阐明(例如肠球菌属对达托霉素的耐药性),耐药机制研究的空白限制了从基因型向表型的推断。
ARGs的表达
细菌对抗菌药物的耐药性不仅仅是由ARGs或特定突变获得驱动的,还受到基因的表达水平的影响。例如,大肠埃希菌含有编码头孢菌素酶的染色体AmpC基因,但由于上游减毒环和弱启动子,该基因在大多数大肠埃希菌中不表达,但启动子或减毒环的突变可导致增加头孢菌素酶的表达,从而增加对青霉素和头孢菌素的可变耐药性。另一个挑战是基因组中ARG的拷贝数,对于一些长序列的基因岛而言,可能需要较长的读长进行测序(例如大量的blaTEM-1导致耐药表型)。
转录组学和机器学习对耐药性预测的贡献
Khaledi等人使用机器学习和转录组分析的方式,依据铜绿假单胞菌的基因型推断其表型,综合考虑ARG的存在/不存在、单核苷酸多态性(SNP)和基因表达时,预测性能会有所提高。
Van Camp等人利用机器学习NCBI生物样本数据库中鲍曼不动杆菌、阴沟肠杆菌、大肠埃希菌、产气肠杆菌和肺炎克雷伯菌共计的915个菌株的表型及ARGs。该模型提供了置信度分数,置信度得分越高,模型的整体性能越好。然而,该模型对氟喹诺酮类药物表现不佳。
ARG与细菌种属
尽管通过一些固有的或特定的ARGs可以很容易鉴定细菌的种属(例如通过mecA鉴定出葡萄球菌),但许多ARGs位于可交换的移动元件上(例如blaCTX-M位于大肠埃希菌和肺炎克雷伯菌等肠杆菌共有的质粒上)。因此,当在宏基因组数据集中同时检测到blaCTX-M基因以及大肠埃希菌和肺炎克雷伯菌时,难以确定该基因具体存在于哪一种菌。
一种可能的解决方案是使用Hi-C交联,即在提取DNA之前将细菌细胞中染色体DNA与可移动的遗传元件(如质粒)进行交联,生物信息学分析就可以将来自位于移动遗传元件上的ARG定位到其宿主的染色体。但Hi-C测序技术是劳动密集型的,且从临床治疗的角度将ARGs和细菌种属相联系的附加价值是有争议的。
因此,从宏基因组数据推断抗菌药物敏感性仍然面临许多挑战。目前,在ARG数据库和相关表型数据的质量方面已经取得了实质性进展,基因型到表型的推断对于肠杆菌、金黄色葡萄球菌和结核分枝杆菌等病原体表现出色。因此,如果可以从宏基因组数据中重建细菌基因组,依据CMg推断抗菌药物敏感性或将成为现实。然而,涉及基因表达的耐药性推断仍具挑战性,仅宏基因组测序可能还不够。未来的研究方向应为通过转录组学和机器学习相结合的方式进一步完善CMg的应用。
点评
传统的药物敏感性判断受限于细菌的长时间培养,相对而言临床宏基因组学在时间上具有相当大的优势。后者可以在若干小时内对细菌敏感性进行评估,有助于指导临床用药决策。然而目前临床宏基因组学判断抗菌药物的敏感性还具有一些挑战,需要进一步联合多组学和机器学习等技术不断完善。
参考文献:Ruppé E, d'Humières C, Armand-Lefèvre L. Inferring antibiotic susceptibility from metagenomic data: dream or reality? Clin Microbiol Infect. 2022 May 9:S1198-743X(22)00229-4.
摘译:曲星伊
审校:李颖
本文转发自华山抗生素所
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017