


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




单位:福建医科大学附属协和医院内分泌代谢科
患者,男性,17岁,体检发现“血钾升高”10天。
患者入院前10天因拟行“双眼斜视”矫正手术于外院术前检查示:“血钾:6.4 mmol/L”,进行补液、利尿等治疗(具体不详)后复查“血钾:5.92 mmol/L;血气:pH 7.34,实际碳酸氢根:17.8 mmol/L”,无多尿、烦渴、四肢无力、胸闷、呼吸困难等不适。
今为进一步诊治,门诊拟“高钾血症原因待查”收住入院。患者自发病以来,精神、睡眠、食欲尚可,二便如常,近1年体重无变化。
出生时体重不详,既往自觉身高较同龄人偏矮;发育正常、智力情况大致正常。5年前曾因身材矮小于外院接受“生长激素”治疗(具体不详)半年,后自行停药。家族中无发现高钾血症。父亲患有“高血压病、甲状腺功能亢进症”,病史1年,余家族史无特殊。
体温(T):37.0℃,脉搏(P):73次/分,呼吸(R):17次/分,血压(BP):129/80 mmHg。身高:157 cm,体重:46 kg,体质指数(BMI)18.6 kg/m2。
神志清楚,体型消瘦,双肺呼吸音清,未闻及干湿性啰音。心率73次/分,律齐,未闻及病理性杂音。全腹软,无压痛及反跳痛,肝脏右肋下、剑突下未触及。双肾未触及,双侧肾区无叩击痛,未闻及血管杂音。肠鸣音正常,4次/分,双下肢无浮肿。外生殖器发育正常。
血常规:白细胞计数7.09×109/L,中性粒细胞68.3%,血红蛋白149.0 g/L,血小板196×109/L。
尿常规:比重 1.032,pH 6.0。
生化全套:钾5.67 mmol/L,钠141.6 mmol/L,氯109.8 mmol/L,钙2.28 mmol/L,镁0.81 mmol/L,磷1.70 mmol/L,碳酸氢盐 19.2mmol/L,阴离子间隙 18,渗透压291 mOSM。
血气分析:pH 7.311,碳酸氢离子浓度18.4 mmol/L,标准碳酸氢根19.0 mmol/L,血乳酸:1.05mmol/L。
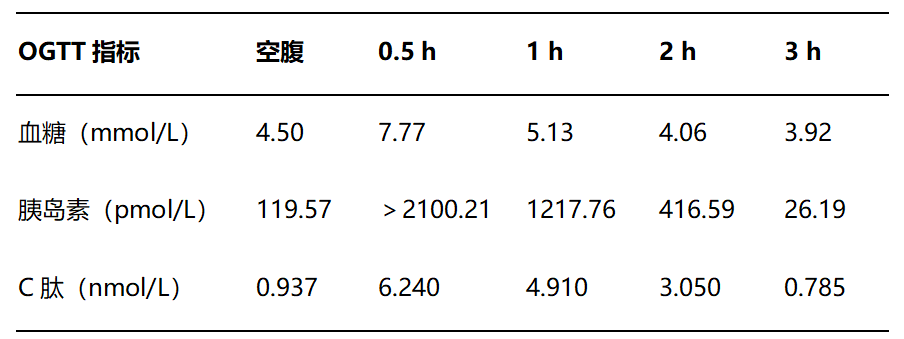
口服葡萄糖耐量试验(OGTT)
24 h尿液检查:尿钾:9.9 mmol/24 h,尿钠:146.3 mmol/24 h,尿钙:3.6 mmol/24 h,尿镁:2.85 mmol/24 h;24 h尿量:1.5 L。
促肾上腺皮质激素(ACTH):35.04 pg/ml(7.2~63.3)。
皮质醇昼夜节律测定(nmol/L):(08:00)537.90、(16:00)293.00、(00:00)271.30。
立位肾素、醛固酮测定:醛固酮11.50 ng/dl(0.0~35.3 ng/dl),肾素4.202 μIU/ml(4.4~46.1 μIU/ml),醛固酮/肾素比值(ARR)2.74,24 h尿醛固酮:1.18 μg/24 h(1.19~28.1)。
游离三碘甲状腺原氨酸(FT3)、游离甲状腺素(FT4)和促甲状腺激素(TSH)正常;性激素正常;生长激素未测。
骨代谢检查:总Ⅰ型前胶原氨基端肽(TP1NP)666.6 ng/ml(9.06~76.24 ng/ml)、Ⅰ型胶原羧基端肽β特殊序列(β-CTX)、骨钙素、甲状旁腺激素正常。
24 h动态血压:全天24 h平均血压138/82 mmHg,白天平均血压142/85 mmHg,夜间平均血压115/64 mmHg,全天血压趋势呈杓型血压。
腹部彩色超声:肝胆胰脾、双肾上腺区、双肾、双输尿管、膀胱、前列腺、双侧精囊腺、门静脉系,腹膜后未见明显异常声像。
心脏彩色超声:房室大小及左室射血分数值正常。
双侧肾上腺CT平扫+增强:双侧肾上腺未见明显异常。
骨龄片:腕关节左手骨龄片相当于16~17岁;骨密度检查未测。
本病例的主要特点为:
年轻男性,肾功能正常的持续性高钾血症,24 h尿电解质提示尿钾排出减少;
高血压;
阴离子间隙正常的代谢性酸中毒;
血醛固酮分泌相对减少。
结合患者临床特点及辅助检查结果,应遵循的临床诊断思路为肾功能正常的高钾血症的诊断与鉴别诊断。
👇👇
患者多次查血钾高,但无明显急性高钾血症症状
考虑持续性高钾血症的病因可能如下
可见于采血时发生血管内溶血、标本储藏不当引起溶血、剧烈运动后即刻采集样本,以及较为少见的情形,如白血病或淋巴瘤等血液病引起标本中血细胞破坏等。
为了维持细胞电中性,钾离子向细胞外转移,此患者虽有代谢性酸中毒,但尿pH值不低,尿钾排泄减少,考虑酸中毒非高钾血症诱因。
患者OGTT及血渗透压结果均不支持。
包括创伤、肿瘤溶解综合征、重度意外性低体温,均与患者病史不符。
通常伴有乳酸升高,与患者检查结果不符。
是一种常染色体显性遗传病,常表现为无力或麻痹,多有受凉、运动后休息、禁食或摄入少量钾等诱因,与患者情况不符。
这些均不符合本例患者病史及检查结果,且患者尿钾排泄减少,故高钾血症的病因考虑肾性钾排泄减少。
急性肾脏病多合并细胞内钾离子释放增加,而慢性肾脏病多合并尿少、高钾饮食或晚期肾衰,均与患者情况不符。
因消化道或肾脏失水,抑或心衰和晚期肝硬化导致组织容量减少,在心衰患者中还可能因使用血管紧张素转化酶抑制剂(ACEI)类药物诱发高钾血症,但本例患者心功能及肝功能均正常,可排除。
低肾素型低醛固酮血症:可见于糖尿病肾病、非甾体抗炎药、急性肾小球肾炎引起的急性容量扩张。
药物:可见于ACEI、直接肾素抑制剂和肝素。
原发性肾上腺皮质功能减退或危重症状态。
以上均与患者病史和表现不符。
先天性孤立性低醛固酮症:① 醛固酮合成酶(P450c11as)基因缺陷,多于儿童起病,特点是反复低血容量、低钠及生长迟滞;② 21-羟化酶(P450c21)基因缺陷,导致皮质醇低下伴不同程度男性化,ACTH升高;均与患者不符。
2型假性低醛固酮血症(PHA2):罕见的单基因遗传病,以肾功能正常的高血钾性高血压和代谢性酸中毒为主要临床表现,病变影响噻嗪敏感性Na-Cl协同转运体(NCC),导致远端小管对氯化钠(NaCl)重吸收增加,输送到皮质集合管泌钾、泌氢细胞的水、钠减少,从而减少了氢、钾排泄。与本例患者的表现较为符合。
例如部分狼疮性肾炎、急性移植排斥反应、接受环孢素或他克莫司治疗、输尿管空肠吻合术后等,这些原因均与患者的病史与表现不符。
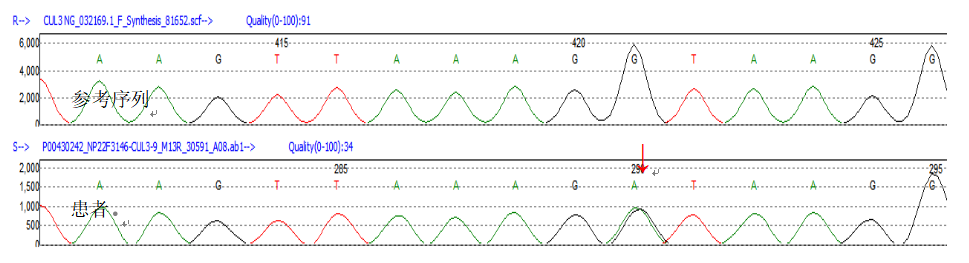
2型假性低醛固酮血症(PHA2),又名Gordon综合征。采集患者外周血送检基因测序,发现CUL3杂合突变(2q36.2 Intron9 c.1377+1G>A)为致病突变(见图1),患者的父母亲均未检测到此致病突变。
图1 本例患者Sanger测序峰图,红色箭头所示为患者CUL3基因c.1377+1G>A突变
予患者口服氢氯噻嗪25 mg qd治疗,3天后复查示血钾和血pH正常。出院后以氢氯噻嗪12.5~25 mg qd维持治疗,复查血钾4.5~4.8 mmol/L,血压范围130~120/90~80 mmHg。
2型假性低醛固酮血症(PHA2),又名Gordon综合征、家族性高血钾性高血压(FHHt),是一种罕见的单基因遗传病(MIM #145260),发病率不明。
临床特点:高血压、高血钾、代谢性酸中毒(阴离子间隙正常)、肾功能正常、醛固酮或血浆肾素活性(PRA)偏低或处于正常低值,可能有家族史,还可伴有身材矮小。
分子缺陷包括两种类型:丝氨酸激酶(WNK1)和苏氨酸激酶(WNK4)异常;降解WNK1[Cullin-3(CUL3)]和WNK4[Kelch-3(KLHL3)]的蛋白突变,主要影响NCC对Na+的重吸收增加。
因此,PHA2临床表现与NCC病变导致NaCl重吸收障碍的Gitelman综合征互为镜像。NCC为噻嗪类利尿剂的作用靶点,因此,治疗核心为限制钠和(或)钾盐摄入联合噻嗪类利尿药,剂量一般选择能将血压降至正常的最小剂量。
电解质紊乱是内分泌科常见疾病,但其病因的诊断往往需要结合临床表现和各项检查结果,抽丝剥茧,追本溯源。此病虽然罕见,但临床工作中仍需要对其提高警惕,尤其是肾功能正常的早发高血压合并高钾的人群,或生长发育异常的青少年,需要尽量明确病因,及早干预,预防高钾血症可能造成的心脏骤停,尽可能减少其他相关并发症的发生,改善预后。

欢迎您参与“疑难病例诊疗提升”专栏互动!无论您想点评病例还是发表学习感悟,抑或对病例诊治仍有困惑,都可以扫描上方二维码给小编留言!
我们会定期精选并刊登读者互动内容,并提供精美小礼品以资鼓励,期待您的积极参与和分享,您的专业视角将为专栏增添更多学术价值与意义!


版权说明:本文系中国医学论坛报社内分泌学科编委会精心出品,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载请联系【中国医学论坛报今日内分泌】申请授权
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017