


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:安徽中医药大学神经病学研究所附属医院 王共强
导读:1872 年乔治·亨廷顿 (George Huntington) 首次描述了遗传性舞蹈病的遗传性质、相关的精神和认知症状以及该疾病在 30 至 40 岁之间的成年生活中的表现。这种舞蹈病现在被称为亨廷顿病 (Huntington's disease,HD)。HD 的单基因性质和完全外显率使其可能成为最可治疗的神经退行性疾病之一。在过去十年中,随着可以直接靶向 HD 基因并阻止有毒突变亨廷顿蛋白 2 产生等基因治疗新治疗方法的出现,为未来带来新希望。
亨廷顿病是一种常染色体显性遗传性神经退行性疾病,除了精神症状和认知改变,还伴有不自主运动,主要是舞蹈运动。在西方人群中,HD 的患病率为每 10 万人中 10.6-13.7 人,常发于40~50岁人群。
HD主要病因是患者第4号染色体上的huntington基因中显性遗传的 CAG 三核苷酸重复扩增引起。在细胞水平上,突变亨廷顿蛋白通过多种机制导致神经元功能障碍和死亡,包括蛋白质稳态、转录和线粒体功能的破坏以及突变蛋白的直接毒性。突变的蛋白质mHTT,mHTT在细胞内逐渐聚集在一起,形成大的分子团,在脑中积聚,损害神经细胞功能。随着疾病的进展,在皮层受累的纹状体中可见早期的宏观变化。这是由于三个DNA构件(CAG)的基因重复次数过多而引起的。字母CAG代表了胞嘧啶,腺嘌呤和鸟嘌呤的缩写。HTT基因中40个以上的这些重复序列会导致脑退行性疾病,最终致死。重复次数越多,症状就越早出现,包括行为障碍,运动和平衡困难,虚弱以及说话和进食困难。
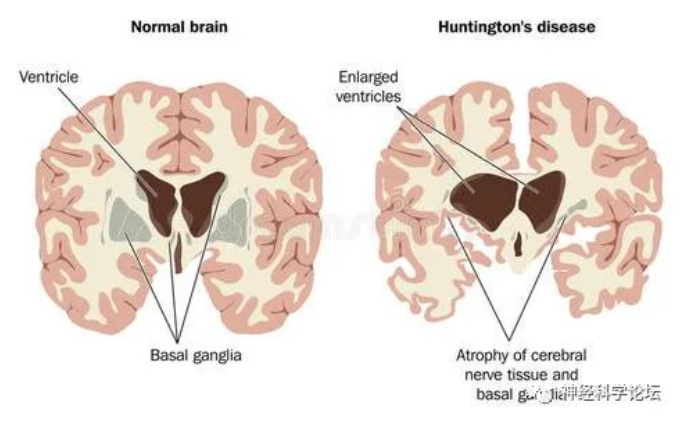
HD一般在中年发病,表现为舞蹈样动作,随着病情进展逐渐丧失说话、行动、思考和吞咽的能力。由于HD影响大脑的不同部位,临床表现特征是神经精神症状、运动障碍(最常见的舞蹈症)和进行性认知障碍,最终将需要全职护理。病情大约会持续发展10年到20年,并最终导致患者死亡。
早期诊断HD对控制病情十分重要。通过在具有该病临床特征的患者中鉴定亨廷顿基因中增加的 CAG 重复长度来确认诊断。尽管诊断通常很简单,但可能会出现不寻常的表现,并且很难知道某人何时从无症状携带者转变为疾病状态。越来越多的情况可以模仿 HD,包括罕见的遗传原因,在 HD 基因检测阴性的情况下必须考虑这些原因。
采用不同放射性显像剂均可敏感显示病灶,多数显像剂主要特异性针对大脑代谢异常、DA通路异常、突触密度下降及脑部炎性反应过程等。HD 早期症状大多特异性不高,影像学表现虽明显但不典型,加之临床病例数少等因素导致诊断HD存在一定困难。不同机制显像对诊断HD具有提示作用。PET是目前诊断HD的最重要的影像学方法之一。
在鉴别诊断上,小舞蹈病常见于5~15岁女性,有风湿病史,亚急性起病,有舞蹈样动作及肌张力减低,病程具有自限性。良性家族性舞蹈病常见于婴幼儿,症状无恶化,不伴有行为人格改变,不伴有
HD目前没有改善疾病的有效治疗方法,支持性和对症治疗是主要治疗方法。主要针对舞蹈症和神经行为问题,尽管支持这些的试验证据通常是有限的。目前普遍的观点是哺乳动物的成年神经元是不可替代的,因此脑疾病的治疗方法往往侧重于减缓疾病的进展。
近十年来,在了解细胞病理学和随着疾病进展而发生的宏观大脑结构变化方面取得了重大进展。HD 的单基因性质和完全外显率使其可能成为最可治疗的神经退行性疾病之一。HD分子发病机制复杂,毒性来源于全长扩展亨廷顿蛋白和亨廷顿蛋白的N端片段,两者都容易因蛋白水解而发生错误折叠;HTT基因的异常内含子1剪接;和 HTT 基因中 CAG 重复序列的体细胞扩增。亨廷顿病的潜在干预措施包括针对亨廷顿 DNA 和 RNA 的疗法、亨廷顿蛋白的清除、DNA 修复途径以及针对炎症和细胞替代的其他治疗策略。随着可以直接靶向 HD 基因并阻止有毒突变亨廷顿蛋白 2 产生的新治疗方法的出现,这一点变得尤为明显,部分疾病改善疗法正在进入试验阶段。但目前看,反义寡核苷酸亚临床试验的提前终止表明未来的基因治疗仍任重道远。
来源:神经科学论坛
罕见病诊疗指南——抗 LGI1 抗体相关脑炎【神经系统罕见病】
《中国卒中学会关于无症状性颈动脉狭窄筛查的科学声明》在线发布
ISC 2022前沿速递|应用西洛他唑双联抗血小板治疗对卒中二级预防影响的性别差异
ISC 2022|续写新篇章 —— “替奈普酶”精彩继续……
血栓预防及血栓形成的治疗推荐丨2022 AHA/ASA自发性脑出血患者管理指南
院前和初始医疗系统的推荐丨2022 AHA/ASA自发性脑出血患者管理指南
Lancet:静脉溶栓替奈普酶(0.25 mg/kg) VS 阿替普酶
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017