


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




患者为男性,现年68岁,65岁时在比利时安特卫普大学医院神经肌肉疾病中心首次就诊。其主诉从40岁起出现非常缓慢进展的肌无力,初表现为行走困难,后逐渐出现右上肢无力,并伴有轻度面肌无力,夜间入睡时双眼不能完全闭合。最初并未提及其20岁时曾被诊断为肺结节病(因症状轻微未予治疗)。
神经系统查体发现患者面部轻度无力,包括双侧轻度上睑下垂及眼轮匝肌无力,右侧近端上肢肩胛带肌肉无力,右侧肩胛骨呈翅状突出。脊柱旁肌、双侧下肢近端肌及足背屈肌均有不同程度无力,但深部指屈肌力完全正常。踝部振动觉消失,胫骨平台及手腕处振动觉减退,其余感觉正常。右上肢及左侧跟腱反射消失,其他腱反射减弱。患者因臀肌无力,步态表现为特征性Trendelenburg步态(因髋部外展肌如臀中肌无力,导致单脚支撑时骨盆下垂,患者为平衡而躯干向支撑侧倾斜,行走时姿态明显异常),并有明显腰椎前凸。血清肌酸激酶稍高(200 U/L,正常参考值25–195 U/L)。
由于患者症状进展极其缓慢,且肌无力分布不对称,临床最初高度怀疑遗传性肌病,尤其是面肩肱型肌营养不良(FSHD)。神经传导检查正常。针极肌电图未见受累肌群有异常自发电位,但在自愿收缩时发现多处肌肉(左下肢近端、腰椎旁、右上肢和肩胛肌群)运动单位电位出现时程延长、高波幅、多相位,提示慢性神经源性改变,但与临床表现并不完全相符。肌肉MRI检查显示脊柱旁、下肢和右侧肩胛肌群有斑片状脂肪变,分布明显不对称,STIR序列未见显著肌肉水肿,这些特征容易被误认为肌营养不良。
进一步对患者进行了分子遗传学检测,包括FSHD 1型和2型,结果均为阴性,外显子测序(WES)亦未发现致病性变异。随后对临床及影像受累明显的右胫前肌进行了肌肉活检,显示肌肉组织广泛肉芽肿性炎症浸润,呈慢性过程,有中度脂肪浸润和纤维化,未见明显肌纤维坏死。免疫组化显示MHC-I在肌膜明显上调,肉芽肿细胞绝大多数为CD68阳性。遗传性肌病相关的各类肌膜蛋白(如dystrophin, α-,β-,γ-sarcoglycans, α-,β-dystroglycan, caveolin 3和telethonin等)表达均正常。综合临床表现、既往肺结节病史及阴性的遗传检测,最终诊断为慢性结节病性肌病。
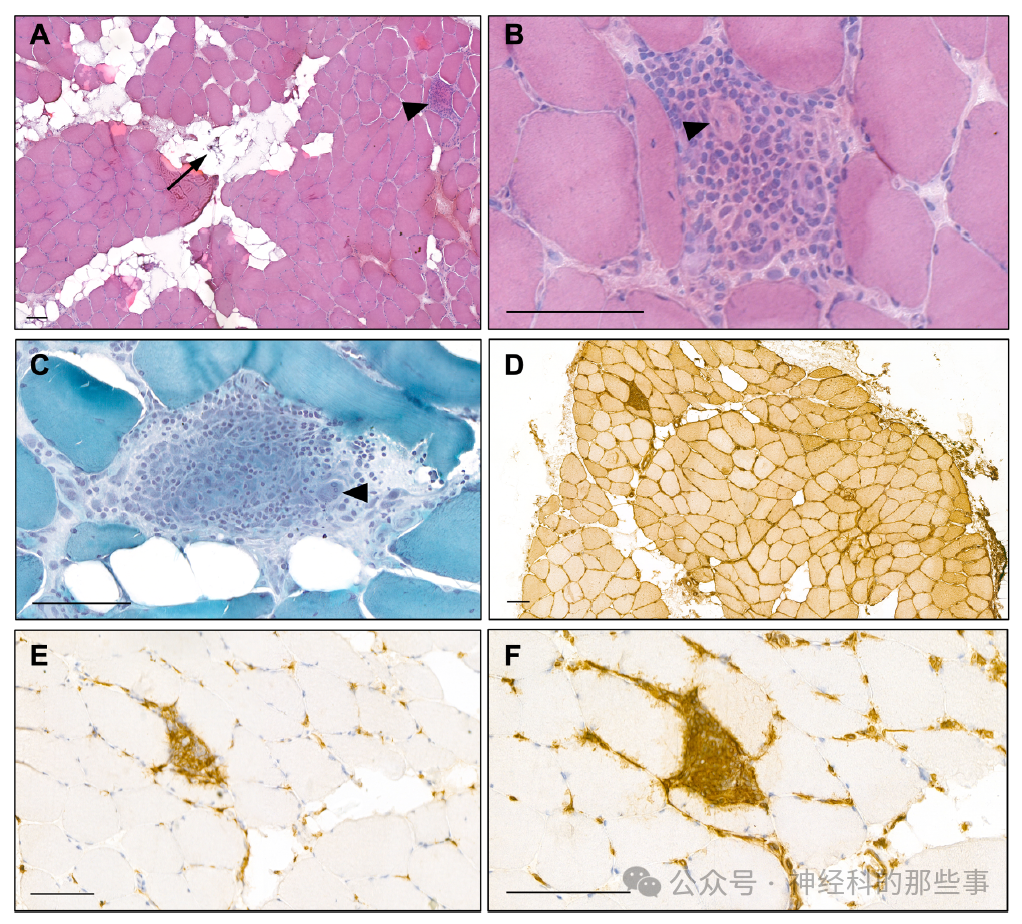
图:(A) 苏木精-伊红(H&E)染色显示肌病特征,可见大量炎性细胞浸润形成非坏死性肉芽肿(箭头),中度肌束膜周围脂肪浸润(箭头),以及轻度肌束内纤维化。(B, C)高倍镜下观察H&E染色(B)和Gomori三色染色(C)下的多核巨细胞(箭头)。(D) MHC-I在肌膜强烈上调。(E) 大多数炎性细胞是CD68免疫反应性组织细胞。(F) MHC-II染色未显示肌膜上调,但突出了含免疫反应性组织细胞和活化T细胞的肉芽肿性浸润。
在全身评估中,18FDG-PET未见“虎人征”(是指在PET中因肌肉受广泛炎症或肉芽肿病变累及而呈现对称性、条纹状、高摄取区,形如老虎皮纹路的一种影像表现,常见于结节病性肌病和其他广泛性肉芽肿性肌病,但并非所有患者都会出现这一征象)。心脏MRI提示左心室下外侧壁呈非缺血性中层延迟强化,考虑可能有心脏受累。由于疾病进展缓慢,未采用糖皮质激素治疗,最初使用甲氨蝶呤,肌无力症状稳定,后因转氨酶升高改为吗替麦考酚酯,患者病情随访两年未见明显进展。复查肌电图及肌肉活检均未发现神经源性损害。
慢性结节病性肌病可表现为极为缓慢进展、临床及影像呈现多灶性、分布不对称的肌无力,极易与遗传性肌病混淆。需要注意的是,与经典文献描述的对称性肢带肌无力不同,慢性结节病性肌病在临床、影像上更常见多灶斑片分布,且深部肌肉(如指屈肌)往往保留。在影像表现中,T1加权序列下可见明显脂肪化,但肌力往往保留较好,提示疾病进展极为缓慢。在鉴别诊断中,除FSHD外,还应注意包涵体肌炎、Caveolin蛋白病、LGMD R2及R12、VCP相关肌病、FHL1相关肌病、DMD或XLMTM女性携带者等。
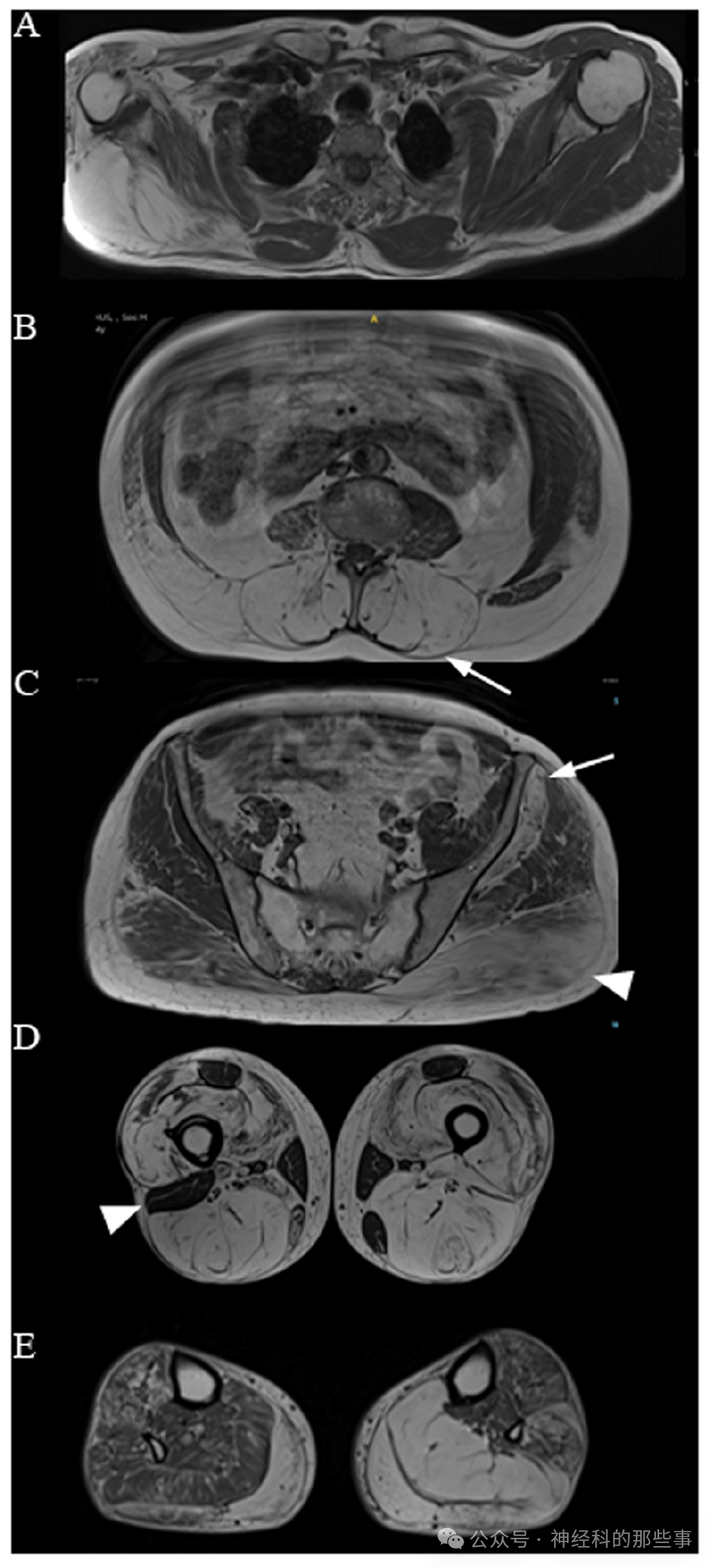
图:轴位T1加权图像分别显示肩部(A)、腹部(B)、骨盆(C)、大腿(D)和小腿(E)水平的图像,呈现出显著的选择性肌肉受累模式:(A)右侧所有肩带肌肉和肩胛周围肌肉,同时累及胸大肌(箭头);(B)腰椎水平的椎旁肌;(C)左侧臀小肌(箭头)和臀大肌(箭头);(D)大部分大腿肌肉受累,但股直肌、缝匠肌、股薄肌和左侧长收肌(箭头)选择性未受累;(E)小腿后侧肌肉受累,前侧小腿肌肉也有较轻程度的受累。
肌肉活检在本病诊断中至关重要,活检部位建议选择临床及影像受累明显的肌肉,以提高诊断率。肌肉活检发现多灶性、非坏死性肉芽肿性炎症,且肌膜蛋白免疫组化无异常,对排除遗传性肌病尤为重要。
在临床实践中,若遇到无法用遗传病因完全解释的多灶性斑片分布的肌无力,应注意排除本病,及时行肌肉活检明确诊断。尽管疾病进展缓慢,部分患者肌无力不明显,但病程过长仍可导致部分器官受累(如心脏),需定期全身评估。治疗优先选择糖皮质激素及免疫抑制剂(如甲氨蝶呤、吗替麦考酚酯等),多数患者可获得症状缓解或稳定。用药期间要密切监测肝肾功能等不良反应并适时调整方案。
Chronic sarcoid myopathy mimicking facioscapulohumeral muscular dystrophy: a case report. Neuromuscul Disord. 2025 Apr 11:50:105364. doi: 10.1016/j.nmd.2025.105364.
来源:神经科的那些事
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017