


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




安 玉 张昌明 梁丹丹 高二志 倪雪峰 程东瑞 左 科 刘志红
[基金项目] 国家自然科学基金专项项目(32141004)
[作者单位] 东部战区总医院 国家肾脏疾病临床医学研究中心(南京,210016)
[通信作者] 刘志红(E-mail:liuzhihong@nju.edu.cn)
DOI:10.3969/j.issn.1006-298X.2023.06.004
摘 要
目的:探讨辅酶Q8B(COQ8B)基因缺陷相关肾病的临床病理及基因变异特征。
方法:回顾分析国家肾脏疾病临床医学研究中心经基因检测诊断COQ8B基因缺陷相关肾病患者的临床资料和家系基因检测结果,总结其临床特点、治疗及预后。
结果:共纳入9例携带COQ8B双等位基因突变的患者,其中2例纯合突变,7例复合杂合突变,所有患者遗传模式符合常染色体隐性遗传特点。基因分析检测到COQ8B基因的7个突变(NM_024876 c.748G>C、c.737G>A、c.1093C>G、c.532C>T、c.449G>A、c.1468C>T、c.1465C>T),其中c.737G>A错义突变为本组患者的热点突变。患者临床表型多局限于肾脏,7例患者以非肾病范围蛋白尿起病,2例表现为肾病综合征,中位起病年龄17岁。5例患者进展至终末期肾病(ESKD),ESKD中位年龄19岁。5例患者影像学提示肾髓质钙化。6例行肾活检的患者中4例表现为局灶节段性肾小球硬化,电镜下可见足细胞和肾小管上皮细胞内线粒体异常。患者对免疫抑制剂治疗无反应。有2例患者采用辅酶Q10治疗,效果欠佳,可能与治疗开始时机较晚有关。
结论:COQ8B基因缺陷相关肾病以青少年期起病的蛋白尿为主要表现,肾脏病理以局灶节段性肾小球硬化为主。肾髓质钙化和电镜下线粒体异常可作为此类患者行基因检测的临床线索。早期基因诊断并及时补充辅酶Q10治疗可能有助于改善患者预后。
关键词 辅酶Q8B基因 肾病综合征 局灶节段性肾小球硬化 肾髓质钙化
Clinicopathological and genetic features of COQ8B nephropathy
AN Yu, ZHANG Changming, LIANG Dandan, GAO Erzhi, NI Xuefeng, CHENG Dongrui, ZUO Ke, LIU Zhihong
National Clinical Research Center for Kidney Diseases, Jinling Hospital, Nanjing 210016, China
Corresponding author:LIU Zhihong (E-mail:liuzhihong@nju.edu.cn)
ABSTRACT
Objective:To investigate the clinical and genetic features of patients with COQ8B nephropathy.
Methodology:We retrospectively analyzed the clinical and pathological manifestations, treatment and prognosis in patients diagnosed with COQ8B nephropathy by genetic test.
Results:A total of nine patients with biallelic mutations of COQ8B were included, among which two patients carried homozygous mutation, and seven patients carried compound heterozygous mutation. The genetic pattern of all patients was consistent with autosomal recessive inheritance pattern. Mutation analysis revealed seven sequence variants in COQ8B (NM_024876 c.748G>C、c.737G>A、c.1093C>G、c.532C>T、c.449G>A、c.1468C>T、c.1465C>T), among which c.737G>A missense mutation is a hotspot mutation in these patients. Patients showed a largely renal-limited phenotype. Of the nine patients, seven started with non-nephrotic proteinuria, and two presented with nephrotic syndrome. The median age at onset was 17 years. Five patients progressed to end-stage kidney disease with a median age at 19 years. Medullary nephrocalcinosis was detected in five patients. Six patients underwent renal biopsy, among which four showed focal segmental glomerulosclerosis. Electron microscopy examination revealed mitochondrial abnormalities. Patients did not respond to immunosuppressive therapy. Treatment with coenzyme Q10 (COQ10) in two patients didn’t result in a significant clinical improvement, which may be due to the late initiation of treatment.
Conclusion:COQ8B mutation can lead to autosomal recessive genetic disease manifested by adolescent-onset proteinuria. The main pathological feature is focal segmental glomerulosclerosis. Mitochondrial abnormalities under electron microscope and medullary nephrocalcinosis may be clinical indicators for genetic test. Early genetic diagnosis and COQ10 supplementation can improve prognosis in these patients.
Key words COQ8B nephrotic syndrome focal segmental glomerulosclerosis medullary nephrocalcinosis
辅酶Q10(COQ10)是一种广泛存在于人体细胞膜上的脂溶性分子,在线粒体呼吸链和能量代谢中起着重要作用。COQ10缺乏可引起多系统受累,表现为脑病、肌张力障碍、肾脏疾病、视网膜病变和感音神经性耳聋等。一系列参与COQ10生物合成和代谢途径的基因功能缺陷均可引起COQ10缺乏,统称为原发性COQ10缺乏症[1-2]。其中位于人类染色体19q13.1的辅酶Q8B(COQ8B)基因(又名ADCK4)编码参与COQ10生物合成过程的一种非典型激酶,该激酶为足细胞迁移所必需,因而COQ8B基因突变引起的临床表型通常局限于肾脏,表现为激素抵抗性肾病综合征(SRNS)和局灶节段性肾小球硬化(FSGS),也称为COQ8B基因缺陷相关肾病[3]。本研究回顾分析了COQ8B基因缺陷所致肾脏病患者的临床病理和基因变异特征,旨在加深临床医生对该病的认识,提高临床诊疗水平。
研究对象 选取2017年3月至2023年4月在国家肾脏病临床医学研究中心经基因检测明确诊断为COQ8B基因缺陷相关肾病的患者。纳入标准:(1)具有慢性肾脏病(CKD)病史;(2)经全外显子检测或肾病panel测序检出COQ8B双等位基因突变,判断与临床表型高度相关且致病性证据充分;(3)临床及家系资料完整。
研究方法 调阅电子医疗系统,收集临床资料、实验室检查及肾活检病理资料。临床资料包括人口学特征、肾脏病临床特征、肾外表型(包括泌尿系统发育异常、心肌病变、神经系统损害、视力和听力障碍)、既往治疗及转归、家族史等。实验室检查包括尿蛋白定量、尿沉渣红细胞计数、血常规、血生化、影像学检查及基因检查结果。病理资料包括光镜、免疫病理及电镜结果。
相关定义 终末期肾病(ESKD)定义为估算的肾小球滤过率(eGFR)<15 mL/(min·1. 73 m2) 或开始肾脏替代治疗。采用基于血清肌酐(SCr)的CKD-EPI公式计算eGFR并评估CKD分期。采用美国医学遗传学与基因组学会(ACMG)发布的《变异解读指南》进行致病性分析[4]。
统计学方法 采用《SPSS 20.0》完成数据管理和统计学分析,计量资料中服从正态分布的变量以均数±标准差表示,偏态分布的变量以中位数(范围)表示。计数资料以例数表示。
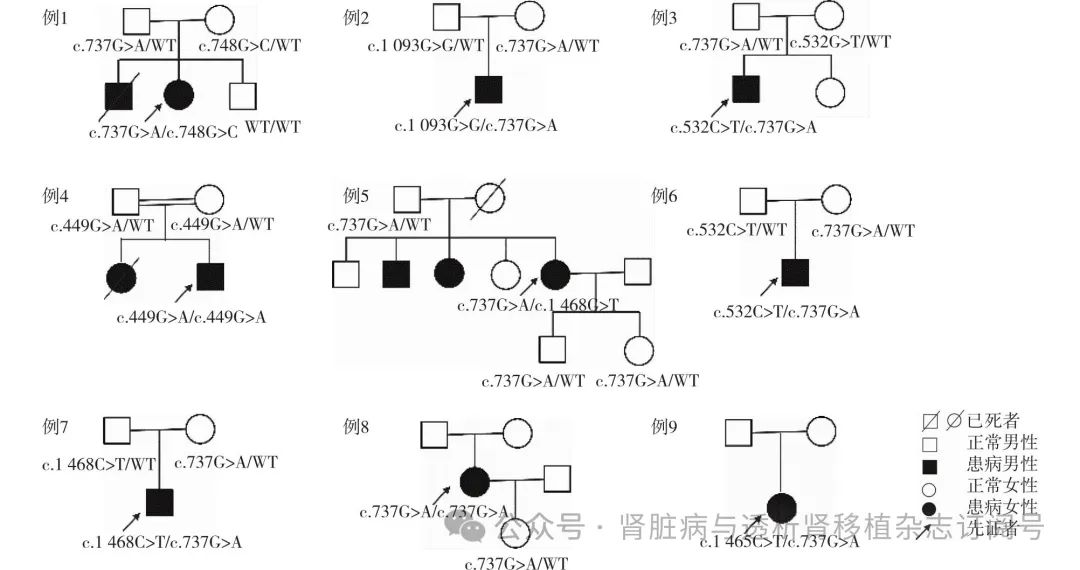
一般情况 研究共纳入9例患者,均为汉族,其中男性5例、女性4例,中位起病年龄为17岁(3~34岁),中位病程为9年(3~17年)。9例患者中有7例(77.8%)起病表现为非肾病范围蛋白尿(≤3.5 g/d),但其中2例(例1和例8)在病程中尿检逐渐加重,进展为肾病综合征(NS)。剩余2例(22.2%)起病即表现NS或SRNS。除1例(11.1%)合并视网膜血管病变外,其余8例均无COQ10缺乏的肾外表现(表1)。3例有肾脏病家族史,其中例1的哥哥两岁出现NS,7岁死于肾衰竭。例4的姐姐亦有蛋白尿,10余岁死于肾衰竭。例5有一哥哥、一姐姐均因肾衰竭目前接受血液透析治疗。
表1 9例COQ8B基因缺陷相关肾病患者的临床特征
ARB:血管紧张素Ⅱ受体拮抗剂;CKD:慢性肾脏病;COQ10:辅酶Q10;ESKD:终末期肾病;FSGS:局灶节段性肾小球硬化;HD:血液透析;MsPGN:系膜增生性肾小球肾炎;ND:未做;NS:肾病综合征;PD:腹膜透析;SRNS:激素抵抗肾病综合征
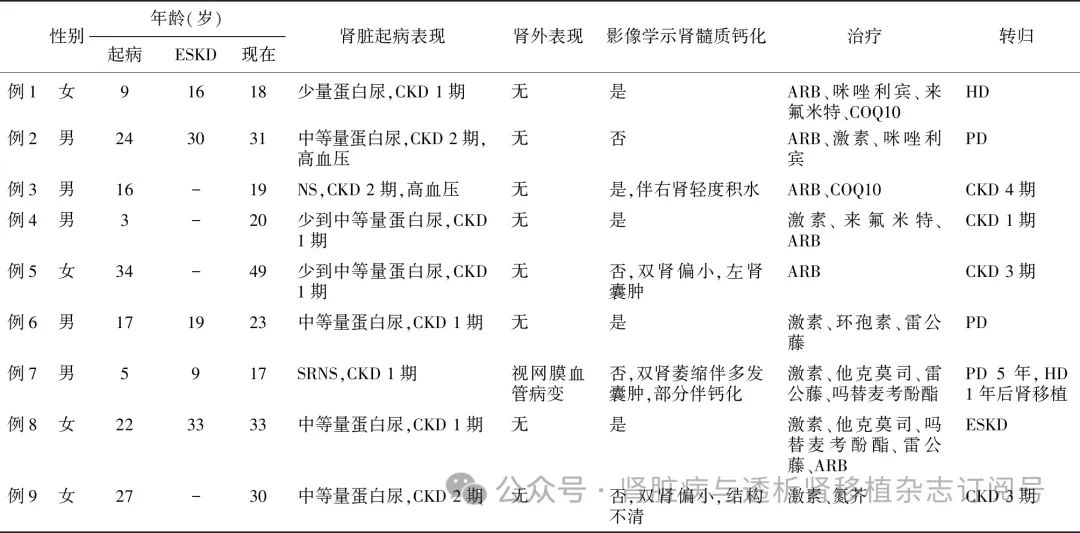
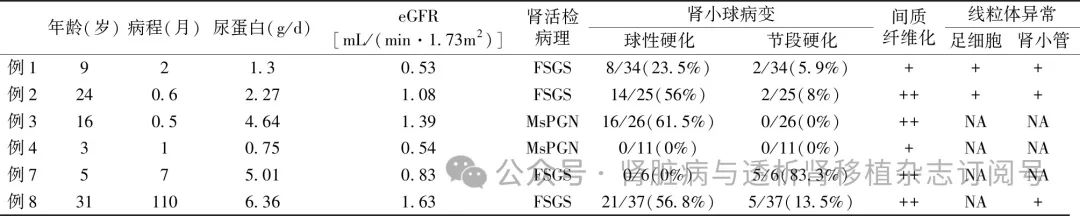
肾脏病理特点 9例患者中6例接受了肾穿刺活检,另3例因肾脏体积缩小或结构不清未行肾活检术。肾活检病理有4例(66.7%)表现为FSGS,这4例患者肾活检时年龄均较轻,但肾脏病理均已表现出不同程度的肾小球硬化和肾小管间质纤维化(表2)。2例(33.3%)表现为系膜增生性肾小球肾炎(MsPGN),例4除MsPGN外,还可见髓质区肾小管呈囊样扩张。6例患者中仅有2例可获得合格的肾活检电镜标本,电镜下除可见足细胞足突广泛融合、消失外,还可见足细胞和肾小管上皮细胞内线粒体数目增多、大小不一,高倍镜下可见线粒体肿胀、嵴紊乱或消失(图1)。
图1 COQ8B基因缺陷相关肾病患者病理改变
A、B:肾小球局灶节段性硬化(PASM-Masson,×400);C、D:肾小球足细胞足突广泛融合、消失(EM);E、F:足细胞足突内线粒体增多,高倍镜下可见线粒体肿胀、嵴消失(EM);G、H:肾小管上皮细胞内线粒体数目增多,大小不一,高倍镜下可见线粒体体积增大、嵴紊乱(EM)
表2 6例COQ8B基因缺陷相关肾病患者肾活检时的临床病理特征
eGFR:估算的肾小球滤过率;CKD:慢性肾脏病;FSGS:局灶节段性肾小球硬化; MsPGN:系膜增生性肾小球肾炎;NA:数据未获得
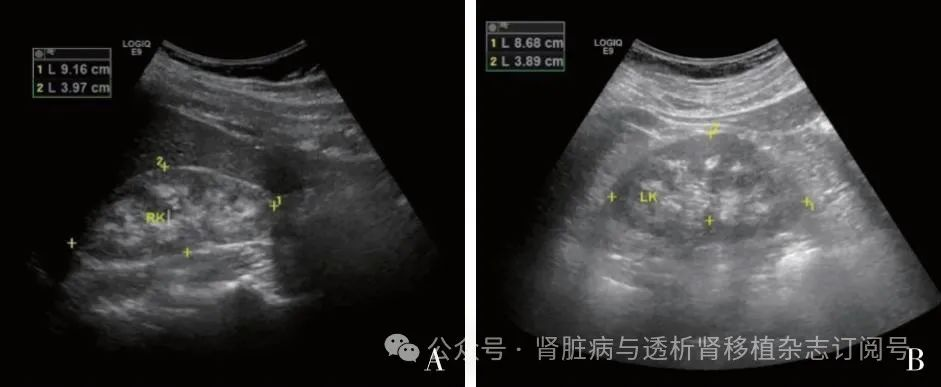
影像学特点 9例患者中有5例肾脏彩超提示双肾髓质回声增强,考虑肾髓质钙化(图2)。例5、例7和例9起病时影像资料已无法获取,现有的泌尿系统彩超提示双肾已萎缩,皮髓分界不清,无法辨识有无肾髓质回声异常。例2泌尿系统彩超未见明显异常。此外,例1曾行CT平扫提示“双肾髓质区多发密度增高影,考虑钙盐沉着可能”,CT尿路造影未见明显异常。例6曾行磁共振检查提示“髓质海绵肾”。
图2 肾脏彩超提示肾髓质回声增强
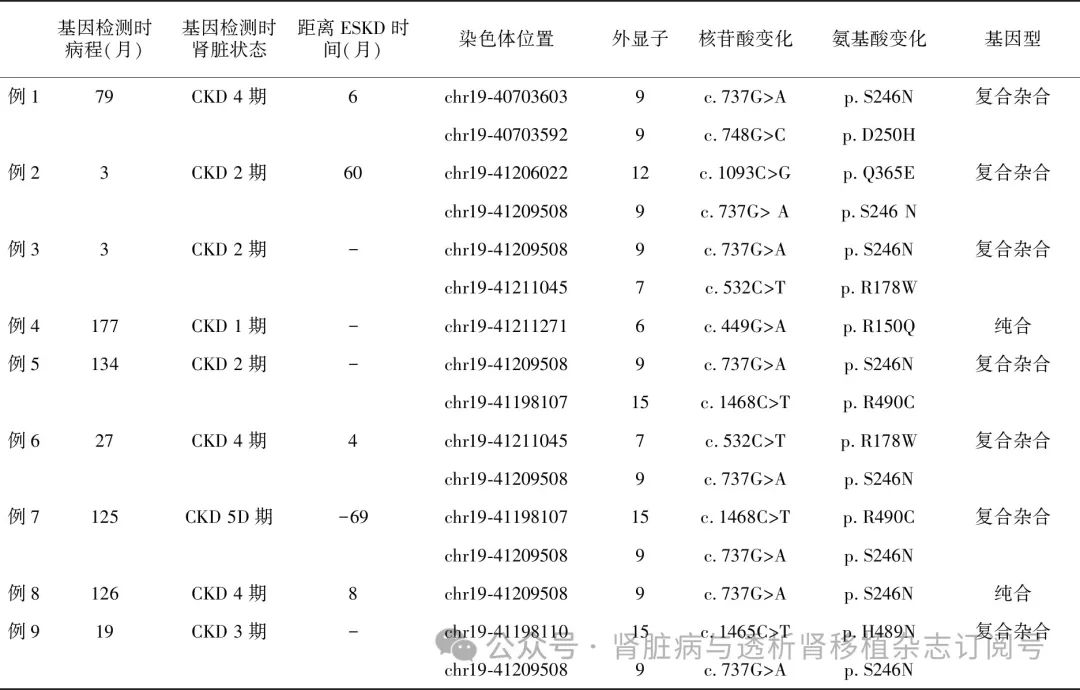
COQ8B基因突变特征 患者行基因检测时的中位病程为79月(3~177月),行基因检测时已有4例患者处于CKD 4~5期,且1例患者已开始肾脏替代治疗。9例患者共检测到COQ8B基因的7个突变(NM_024876 c.748G>C、c.737G>A、c.1093C>G、c.532C>T、c.449G>A、c.1468C>T、c.1465C>T),均为已报道的致病错义突变(表3)。其中例4患者父母系姨表近亲结婚,携带c.449G>A纯合突变,例8为c.737G>A纯合突变,其余7例均为复合杂合突变。c.737G>A位点错义突变在本组患者中发生频率较高(8例),提示其可能为中国汉族人群COQ8B基因缺陷的热点突变。其次则为c.532C>T和c.1468C>T位点错义突变,各2例患者。所有患者遗传模式符合常染色体隐性遗传特点(图3)。
图3 9例COQ8B基因缺陷相关肾病患者家系图
表3 9例COQ8B基因缺陷相关肾病患者基因检测的时机及基因型
CKD:慢性肾脏病;CKD 5D期:慢性肾脏病5期并已行透析治疗;ESKD:终末期肾病
治疗及预后 9例患者中有5例患者已进入ESKD,进入ESKD的中位年龄为19岁(9~33岁),4例患者已开始接受肾脏替代治疗,其中1例为维持性血液透析,2例腹膜透析。例7在腹膜透析治疗5年后因并发腹膜炎,改为血液透析治疗1年后接受同种异体肾移植术,目前肾移植术后2年,尿检和肾功能持续正常。但也有2例患者(例4和例5)病情长期保持稳定,仅少到中等量蛋白尿,提示疾病的发展具有异质性。
7例患者病程中曾使用激素和多种免疫抑制剂治疗,均对治疗无反应(表1)。2例患者(例1和例3)在基因诊断后尝试采用口服COQ10治疗。其中例1启动治疗时的eGFR为19 mL/(min·1.73m2),口服COQ10[10 mg/(kg·d)]治疗3月后复查eGFR为9 mL/(min·1.73m2),仍大量蛋白尿,后患者自行停药。例3启动治疗时的eGFR为56 mL/(min·1.73m2),口服COQ10治疗2年,尿蛋白曾由7.38 g/d降至1.74 g/d,后再次增加,且eGFR仍进行性降低[末次随访时23 mL/(min·1.73m2)]。
COQ8B基因缺陷相关肾病是2013年由Ashraf等[5]首次报道的一种临床表现为SRNS,肾脏病理提示为FSGS的常染色体隐性遗传单基因病,随后国内外陆续报道了100多例患者[6-7]。目前该病的确切发病率尚不清楚,国外研究显示,其在SRNS患者中占0.17%~3.7%,也有数据表明它是引起中国和韩国儿童SRNS较常见的原因之一[8-9]。既往宋晓翔等[10]在69例表现为SRNS或持续性蛋白尿的患儿中检出8例COQ8B基因突变,比例高达11.6%。另一项来自中国儿童遗传性肾脏疾病数据库的分析中1 001例患者共检出16例COQ8B基因缺陷相关肾病患儿,在遗传性肾脏疾病中约占3.8%,SRNS中约占5.7%[11]。
在Ashraf等[5]首次报道的15例患者中,所有患者均表现为SRNS,其中13例行肾活检,12例表现为FSGS。但随后的研究表明,除SRNS和FSGS外,COQ8B基因缺陷相关肾病也可表现为非肾病范围蛋白尿和MsPGN[12-13]。Korkmaz等[12]报道了来自12个家族的26例COQ8B肾病患者,仅44%表现为NS。与其他引起儿童SRNS的常见突变基因如WT1和NPHS2(多在5岁前发病)相比,COQ8B肾病起病年龄更晚,但发病后的肾功能减退速度更快,约80%患者在20岁前进入ESKD,接受肾脏替代治疗的中位年龄为 16.1岁[12]。
最新发表在《Kidney International》上的一项研究纳入了既往文献和数据库调研中的140例COQ8B基因缺陷相关肾病患者进行分析,结果显示该病的中位起病年龄为9.8岁,约71.7%的患者表现为NS,28.3%的患者表现为非肾病范围蛋白尿。病理显示77.1%患者表现为FSGS,15.6%患者表现为球性硬化,7.2%表现为MsPGN。与其他COQ10基因缺陷常引起多系统损害不同的是, 肾外表现仅见于29.3%COQ8B基因突变的患者,其中最常见的是神经系统异常(12.1%),其次为心血管异常(7.1%)、眼底改变(5%)[13]。
本研究中纳入的所有患者均出现肾脏受累,肾外受累比例低,仅1例患者合并视网膜血管病变。与既往研究相比,本组患者发病年龄更晚,其中仅有2例患者起病表现为典型NS,其余7例起病均表现为非肾病范围蛋白尿。虽然病理表现仍以FSGS为主,但也有33.3%表现为MsPGN。值得注意的是,尽管大多数患者在起病后数年内快速进展至ESKD,也有部分患者如例3和例4病情长期保持较为稳定。相较以往研究多来自儿科,本研究显示了COQ8B基因缺陷相关肾病在成人患者临床表现更不典型。由于成人隐匿起病的尿检异常并不常规行遗传学检测,因而该病的实际发病率有待进一步评估。
本研究中9个家系检出COQ8B的7个突变位点,均为错义突变, 其中c.737G>A位点错义突变在本组患者中发生频率较高,这与Song等[14]的研究结果一致,提示其可能为中国汉族人群COQ8B基因缺陷的热点突变。但受样本量限制,本研究未发现突变位点与临床表型之间的关系,可能需要在后续研究中进一步探索。Park等[15]报道了6例病理表现为FSGS的COQ8B基因缺陷相关肾病患儿,所有患者电镜下均可见线粒体异常,影像学均提示肾髓质钙化,推测这种钙质沉着可能与线粒体功能障碍导致肾小管上皮细胞内钙代谢紊乱及氧自由基介导的肾小管上皮细胞损伤有关。在本研究中我们也观察到5例的患者影像学提示肾髓质钙化,电镜下可见足细胞和肾小管上皮细胞线粒体异常。因而线粒体异常和肾髓质钙化可作为此类患者行基因检查的临床线索。
COQ8B基因突变导致肾脏损伤的机制与其影响足细胞中COQ10的生物合成和线粒体功能有关。该基因在大鼠和人类肾脏足细胞线粒体和足突高表达,敲除COQ8B可在斑马鱼和果蝇模型中诱导出NS的临床表型,并可使体外培养的足细胞迁移能力下降,而补充COQ10可逆转这一反应[5]。Widmeier等[16]通过构建足细胞特异性COQ8B敲除的小鼠模型和人足细胞系,进一步证实了COQ8B敲除可导致小鼠出现FSGS和蛋白尿的表型,并导致体外培养的足细胞内COQ10合成减少及线粒体功能障碍,补充2,4-二羟基苯甲酸(一种COQ10前体类似物)可逆转线粒体功能障碍并防止肾损伤进展。因而对此类患者补充COQ10可能是有效的。
既往也有一系列小样本的回顾性研究和个案报道探索了口服COQ10治疗COQ8B基因缺陷相关肾病的疗效,结果显示仅在部分患者可减少蛋白尿,延缓肾脏病进展[14,17-20]。最近Drovandi等[20]汇总了COQ10在原发性COQ10缺乏症患者中的疗效,结果显示,治疗后44%的COQ8B基因缺陷相关肾病的患者蛋白尿下降50%以上,16%的患者蛋白尿完全缓解。在肾功能保护方面,COQ10治疗可延缓eGFR下降的速率。补充和不补充COQ10的患者(每组18例)5年肾脏生存率分别为55% 和29.5%,但二者并无统计学差异(P=0.057 79)。治疗失败的原因可能与启动时机较晚、肾脏病变已进展至不可逆阶段,以及口服COQ10的生物利用度差等有关[16]。
由于COQ8B基因缺陷相关肾病起病隐匿且病情进展较快,许多患者在确诊时已错过治疗的最佳时机。在Korkmaz等[12]的研究中,约46%的患者在诊断时肾功能已进展至CKD 3期以上,26.9%诊断后即开始肾脏替代治疗。Drovandi等[13]报道74.2%的患者在18岁时已进入ESKD,从首次出现症状到ESKD的中位时间仅1年。本组患者中44.4%基因诊断时已处于CKD 4期以上并在基因诊断1年内进入ESKD。尽管有2例患者采用COQ10治疗,但在启动治疗时肾功能均已进展至CKD 3~4期,治疗效果欠佳。因此,早期进行基因诊断并进行针对性治疗是改善此类患者预后的关键,对于家系中无症状成员的筛查更加重要。
小结:COQ8B基因缺陷相关肾病是由COQ8B基因突变引起的以足细胞中COQ10合成减少和线粒体功能障碍为主要表现的一种单基因病。该病多表现为孤立性蛋白尿或SRNS,肾活检病理以FSGS为主。电镜下线粒体异常和影像学提示肾髓质钙化可作为此类患者行基因检查的临床线索。由于该病起病年龄较晚、起病隐匿但进展迅速,早期进行基因诊断并及时补充COQ10治疗有助于改善患者预后。


来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017