


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:东部战区总医院 国家肾脏疾病临床医学研究中心 全军肾脏病研究所 焦晨峰 赵丽丽 程震 刘志红
主诉 患者12岁男童,因“发现蛋白尿4年余,尿糖阳性3月”于2018年12月18日入院。
现病史 2014年6月,患儿母亲发现患儿尿中泡沫增多,遂至南京医科大学附属儿童医院就诊。检查结果显示,患儿尿蛋白阳性(±~++),无镜下血尿,肾功能、血压均正常。后返至当地私人诊所,长期服用中药治疗。曾服用多种中药,包括黄柏、泽泻、猪苓、茯苓、苍木、滑石、白茅根、金银花、蒲公英、玉米须、生苡仁、车前子等。其间曾复查尿蛋白,始终阳性(波动范围+-~++)。2018年9月中旬,患者至当地医院复查,发现尿糖阳性,尿蛋白++。2018年12月3日至我院门诊就诊,检查结果显示,尿糖+,尿蛋白2.21g/24h,尿微量白蛋白319.74mg/24h,尿N-乙酰-β-D-氨基葡萄糖苷酶(NAG)54.8U/(g·cr),尿视黄醇结合蛋白(RBP)38.6mg/L,血肌酐0.44mg/dl,尿酸132μmol/L,空腹血糖4.9mmol/L。现为进一步诊治收治入院。起病以来,患者无不规则发热、皮疹、关节痛或肢端麻木等表现。精神、体力、食欲和睡眠正常,尿量正常,夜尿0~1次,大便正常,体重无明显变化。
既往史 无特殊。
个人史 独生子,生长发育正常,智力正常,余无特殊。
家族史 父母非近亲结婚,父母尿检及肾功能正常,否认家族肾脏疾病和泌尿系结石家族史。
体温36.9℃,心率85次/分,血压110/70mmHg,身高155cm,体重47kg,体质指数(BMI)19.6 kg/m2。神志清楚,全身浅表淋巴结未触及肿大,心、肺及腹部查体未见异常,双下肢不肿。
尿液 尿蛋白定量2.54g/24h,尿白蛋白319.74mg/24h,尿沉渣红细胞阴性,尿pH 6.5,尿NAG27.1U/(g·cr),RBP4.87 mg/L、尿嗜酸细胞计数0.00个/ml,尿糖++。尿α2-巨球蛋白、补体C3正常。尿葡萄糖1.911g/24h、尿钙/尿肌酐比值0.229(正常参考范围:<0.2),尿酸排泄分数17.7%(正常参考范围:7%~12%),尿草酸44.42 mg/24h(正常参考范围:<45mg/24h),枸橼酸209.48mg/24h(正常参考范围:>320mg/24h)。
血液 血常规:白细胞计数6.24×109/L,血红蛋白129g/L,血小板计数232×109/L。C反应蛋白<0.5mg/L,丙氨酸氨基转移酶14 U/L,门冬氨酸氨基转移酶19U/L,白蛋白46.7g/L,球蛋白22.8g/L,尿素氮13.8 mg/dl,肌酐0.54mg/dl,尿酸162 μmol/L,甘油三酯0.71mmol/L,总胆固醇2.86mmol/L,空腹血糖5.06mmol/L,餐后2 小时血糖6.4mmol/L,血钾3.64mmol/L,血钠137.7mmol/L,血镁0.9mmol/L,血钙 2.43mmol/L,血磷1.22mmol/L,总二氧化碳25.9mmol/L,糖化血红蛋白(HbA1c)5.0%。抗核抗体(ANA)、抗双链DNA抗体(A-dsDNA)阴性,抗磷脂酶A2受体(PLA2R)抗体阴性,补体C30.854g/L,补 体C40.187g/L。IgG6.6g/L,IgE46.30 IU/ml,IgM0.954g/L,IgA1.29g/L,类风湿因子(RF)<20.00IU/ml,抗链球菌溶血素(ASO)27.10IU/ml。淋巴细胞亚群:CD4+ T淋巴细胞657个/μl,CD8+T淋巴细胞212个/μl,CD3+T淋巴细胞898个/μl。甲状腺功能正常。
其他 心电图:正常。肾脏B超:左肾102 mm×46mm×51mm,右肾104mm×39 mm×52mm;双肾皮质回声稍增强,内可见数个点状强回声,未见后声影。肾脏B超报告:①双肾皮质回声稍增强;②双肾结晶。肾脏计算机体层摄影(CT):双肾平扫未见异常。
光镜 光镜下观察皮质肾组织1条(图1A~C)。33个肾小球中1个肾小球废弃,个别肾小球发育欠佳,余肾小球系膜区偶见轻度增宽,毛细血管袢开放好,囊壁节段增厚。过碘酸希夫-乌洛托品银-马松(PASM-Masson)染色阴性。肾小管间质病变较轻,部分小管上皮细胞肿胀,偶见小管萎缩、基膜增厚,少数未萎缩小管基膜亦增厚,管腔内少量蛋白管型,间质散在单个核细胞浸润。动脉未见明确病变。
免疫荧光 肾小球12个,冰冻切片荧光染色IgM阳性、补体C3阳性,弥漫分布,呈颗粒状沉积于系膜区。IgG、IgA和补体C1q阴性。小管基膜未见免疫复合物和补体沉积。球门区血管、管间毛细血管及间质血管未见免疫复合物、补体沉积。冰冻切片荧光染色κ轻链、λ轻链均阴性。
电镜 电镜下观察1个肾小球(图1D、E)。肾小球系膜区略增宽,系膜区未见电子致密物沉积。肾小球毛细血管袢开放好,基膜内皮下、上皮侧未见电子致密物沉积。肾小球足细胞足突偶见融合。肾小管上皮细胞胞浆内见较多线粒体,少数线粒体嵴紊乱,小管上皮细胞近基膜侧见内褶明显,个别小管基膜内见囊泡。
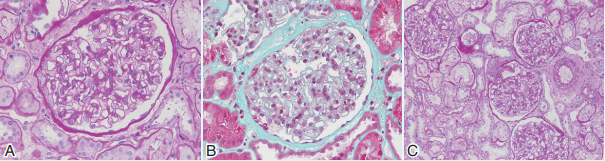
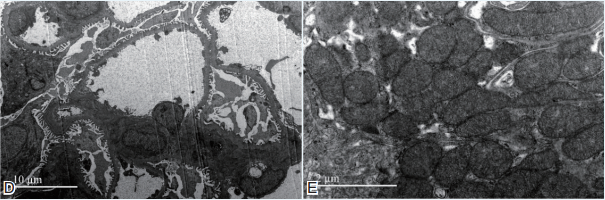
图1 患者肾组织病理改变 A.光镜下肾小球系膜区略增宽,毛细血管袢开放良好,囊壁增厚[过碘酸希夫(PAS)染色,×400];B.肾小球内未见免疫复合物沉积[马松(Masson)染色,×400];C.肾小管间质少许炎细胞浸润(PAS染色,×200);D.肾小球系膜区略增宽,系膜区、基膜内皮下、上皮侧未见电子致密物沉积,足细胞足突偶见融合;E.肾小管上皮细胞胞浆内见少许线粒体嵴紊乱

图2 CLCN5基因检测结果
基因测序检查 基因测序结果显示,该患儿在染色体chrX-49855539位置CLCN5基因11号外显子插入AC两个碱基(c.2146_2147insAC),即编码区第2146和第2147核苷酸之间插入腺嘌呤和胞嘧啶。该插入突变导致移码突变,第717号氨基酸由苏氨酸变为甘氨酸,移码后编译13个氨基酸终止。该患儿为CLCN5基因突变的半合子变异,变异来源于患儿母亲(图2)。
青少年男性,病程4年,起病隐匿,临床表现少到中等量蛋白尿、无镜下血尿、血压及肾功能正常。此外,患儿肾小管损伤指标明显升高,肾性糖尿及低尿酸血症。患者24小时尿微量白蛋白水平与24小时尿蛋白定量比值14.5%,证实患者尿蛋白以非白蛋白为主。肾穿刺活检光镜下病变轻微,电镜偶见足细胞融合。最终考虑患者蛋白尿主要来源于肾小管疾病。进一步完善基因检测后发现患儿CLCN5基因突变。
最终诊断 Dent病1型。
患者明确诊断后,给予血管紧张素转换酶抑制剂(ACEI)、氢氯噻嗪联合枸橼酸钾治疗。末次2021年12月门诊随访复查尿蛋白定量2g/d,尿NAG38.1U/(g·Cr),RBP7.72mg/L,白蛋白45.9g/L,肌酐0.65mg/dl,尿酸157μmol/L,电解质正常。
多种病因均可导致肾小管损伤。根据其导致近端肾小管损伤发病机制,可分为获得性及遗传性疾病。获得性疾病包括服用易导致肾脏损伤的药物(如阿德福韦酯、庆大霉素及中草药)和重金属(如铅、镉)中毒等。该患者病程中曾服用多种中药(不含木通等成分),但患者无发热、皮疹及嗜酸性粒细胞增多等药物过敏表现,且患者起病前即有尿检异常,停用相关药物1年后,尿检异常及肾小管指标未恢复正常,故不支持单纯中药导致肾小管损伤的诊断。遗传性疾病包括胱氨酸尿、半乳糖血症、肝豆状核变性、遗传性果糖不耐受症、糖原贮积病及线粒体病等,但患者临床表现及基因检测结果均不支持诊断为上述疾病。
患儿血钙正常,尿钙排泄升高,肾脏B超提示双肾结晶,多种病因可导致肾脏钙盐沉积。需要与以下疾病鉴别:①饮食摄入钙剂和(或)维生素D制剂增多。但患者病程中无长期高钙饮食,且无长期服用维生素D制剂。②结节病。该病表现为多系统受累,且以肺部及淋巴结最为常见,血钙正常或升高。患儿胸部CT未见异常,全身未触及肿大淋巴结,肾脏活检病理未见典型多核巨细胞性肉芽肿性间质性肾炎。③肾小管酸中毒。该病的临床表现为骨痛、病理性骨折、代谢性酸中毒、反常性碱性尿和尿比重降低,可伴有低钙、低磷及低钾。患儿尿液pH正常,无酸中毒表现。④髓质海绵肾。该病以血尿常见,CT平扫见肾锥体多发斑点状高密度影及扩张接合管形成囊状低密度影。该患儿无血尿,CT平扫未见上述表现。
该患者有低分子蛋白尿、高尿钙及肾结晶,结合基因检测结果和遗传方式,最终诊断该患儿为Dent病1型所致肾小管性蛋白尿。
Dent病是一种罕见的儿童X连锁隐性遗传性肾小管疾病,1964年由登特(Dent)首次报道此类疾病。患者临床表现为低分子蛋白尿、高尿钙、肾钙化和(或)肾结石,可有其他近端肾小管功能异常,逐渐进展至肾功能不全。部分患者可合并佝偻病或骨软化症。该病起病隐匿,表现不典型,临床上常因认识不足而漏诊、误诊。
Dent病多起源于儿童期或青少年期,主要见于男性。大多数男性患儿,尿蛋白定量波动在0.5~2.5g/24h,部分患者可达到4g及以上。其蛋白尿性质仍为肾小管性蛋白尿,低分子蛋白尿占比50%以上,临床通常不引起低蛋白血症、高脂血症和水肿。以上特点可与肾小球疾病导致的蛋白尿相区别。其他临床表现包括肾性糖尿、氨基酸尿、磷酸盐尿、低尿酸血症、低磷血症、佝偻病或骨软化症。女性患者一般临床表现轻微。
对于Dent病,目前国内外学者通常采用以下临床诊断标准:①低分子蛋白尿,即尿蛋白电泳提示低分子蛋白>50%,或尿α1-微球蛋白、β2-微球蛋白和(或)视黄醇结合蛋白升高至少高于正常上限5倍,且以低分子蛋白为主;②高钙尿症,即24h尿钙>4mg/kg(>1mmol/kg)或随机尿钙/尿肌酐比值>0.25;③至少有下列情况之一:肾钙化、肾石症、血尿、低磷血症或肾功能不全。满足上述3条临床诊断标准者方可临床确诊Dent病。也有作者将随机尿钙/尿肌酐比值>0.2作为高钙尿症的诊断标准。本患者符合上述3条临床诊断标准。
Dent病根据其基因突变不同分为2型。Dent病1型与编码氯离子通道蛋白5(CLC-5)的CLCN5基因变异相关,该基因位于染色体Xp11.22位点,全长2238bp。Dent病2型与编码磷脂酰肌醇5-磷酸酶的OCRL2基因变异相关,该基因位于Xq25位点。其中Dent病1型占60%,2型占15%,分子遗传不明者占25%。本例患者完善基因检测提示,CLCN5基因突变,诊断为Dent病1型。目前已报道有250余种CLCN5基因突变,包括错义突变(44%)、无义突变(26%)、剪接位点突变(11%)、小缺失或插入(15%)和大缺失(4%)。CLC-5主要在髓袢升支粗段及集合管闰细胞表达。CLCN5基因突变可导致CLC-5蛋白截断或缺失,进而导致其功能丧失,影响近端肾小管上皮细胞氯离子电导的损耗及逆向转运蛋白功能缺陷,损伤囊泡的酸化功能,造成近端肾小管细胞胞吞作用障碍,从而导致一系列临床异常。
Dent病2型为OCRL2基因突变,临床上需要注意OCRL2基因突变亦可导致Lowe综合征。Lowe综合征临床表现为先天性白内障、智力障碍及近端肾小管功能障碍,与Dent病2型相比,Lowe综合征患者氨基酸尿、糖尿、肾小管酸中毒更为常见,但高尿钙、肾结石和肾钙化少见。
Dent病肾脏病理无特异性改变,早期病理病变轻,随着病变进展,光镜下可见肾小管萎缩、间质纤维化、局灶节段肾小球硬化、球性硬化和系膜增生等改变。免疫荧光通常阴性。电镜下可见足突轻度融合。随着疾病的进展,肾小球硬化、肾小管萎缩、间质纤维化及足突融合程度逐渐加重。本例患者肾脏病理仅有肾小管间质轻度改变,电镜下偶有足突融合。与文献报道一致。
目前Dent病缺乏有效的治疗手段。治疗目标是降低高尿钙、减少肾钙化和肾结石,延缓肾功能不全进展。常规治疗手段包括多饮水、低盐及高柠檬酸饮食、避免高钙及高草酸饮食。对于生长发育儿童,不建议低钙饮食,避免影响骨骼生长发育。治疗上给予氢氯噻嗪,促进远端小管对钙离子重吸收,降低尿钙浓度;联合枸橼酸钾,防治氢氯噻嗪导致低钾血症,同时可碱化尿液、升高尿枸橼酸盐浓度,减少肾钙化、肾结石。目前尚无证据表明给予ACEI或血管紧张素受体阻滞剂(ARB)类药物可以减少蛋白尿,但可能延缓肾小管间质慢性病变。有学者对CLCN5基因敲除的小鼠行骨髓移植,移植术后4月,近端小管功能明显改善。目前骨髓移植尚未应用于Dent病临床治疗,有待进一步评估。
该病预后差,男性Dent病患者中每年肾小球滤过率下降1.0~1.6ml/min。约30%~80%的男性患者在30~50岁进入终末期肾病。50%的患者可发展至肾病范围蛋白尿,但非肾病综合征状态。对于终末期肾病患者,可考虑肾移植手术治疗。
Dent病是一种罕见的X连锁隐性遗传肾小管疾病,对于有低分子蛋白尿和(或)高尿钙伴肾石症的男性患儿,应警惕Dent病,及早完善基因检测有助于诊断,避免误诊和漏诊。治疗上予以积极控制高尿钙,防治肾钙化及肾石症。

来源:《中国医学论坛报》2022年9月15日A4-A5版
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017